
Abstract
Histone proteins are subject to several modifications, including phosphorylation, acetylation, methylation, sumoylation, and ubiquitination. These posttranslational modifications play critical roles in chromatin structure and gene transcription. Because of their involvement in the progression of a variety of diseases, histone modifications are attracting increased attention. We report herein a high-throughput DELFIA assay to quantify H3K27me3 in the prostate cancer cell line, PC3. Using a high binding MaxiSorp plate, we were able to eliminate the need for the capture antibody. We also developed an effective method, a combination of “freeze-thaw” and 0.2 N HCl, to extract histone proteins in PC3 cells cultured in a 384-well plate. To compensate for cell viability change, we normalized H3K27me3 signal to the total amount of H3 in each sample well. As a result, we show that the assay has a good dynamic range with a robust assay window. Using a methlytransferase inhibitor, DZNep, we show that the change of H3K27me3 signal is target specific. This method simplifies the logistics in screening and profiling and reduces the cost per well to an acceptable level for high-throughput screening. The findings presented here should be applicable to other assays involving binding and extraction of histone proteins.
Introduction
The nucleosome is the essential repeating unit of chromatin consisting of 147 bp DNA wrapped around an octamer of the core histone proteins—namely, H2A, H2B, H3, and H4.1,2 In comparison with the C-terminal domains, the N-terminal domains of all histone proteins are less structured and are therefore subject to chemical modifications affecting chromatin structure and gene expression. These posttranslational modifications include acetylation, methylation, ubiquitination, and phosphorylation. 3 Histone H3, one of the core components of the nucleosome, is the most extensively modified histone protein. The methylation of histones can occur on two different residues: arginine and lysine. Lysine 27 of histone H3 (i.e., H3K27) can be mono-, di-, or trimethylated by different histone-lysine-N-methyltransferases such as Enhancer of Zeste homolog 2 (EZH2) or nuclear-receptor set domain (NSD3). Generally, acetylation of lysine residues on histone H3 and H4 opens up chromatin structure, resulting in increased transcription. 4 In contrast, methylation of lysine residues can lead to either activation or repression of gene expression depending on where the methylation occurs. 5 H3K27me3 is involved in the regulation of homeotic (Hox) gene expression and in the early development stage of X-chromosome inactivation. 6 It serves as a hallmark for specific binding of the chromodomain of polycomb repressor complex (PRC1), which includes B lymphoma Mo-MLV insert region 1 (BMI-1), ring finger protein 1 (RING1), human polycomb (HPC), and human polyhomeotic homologue (HPH). 7 The recruitment of PRC1 to H3K27me3 sites halts initiation of transcription by RNA polymerase II, which further represses gene transcription.8,9 H3K4me2/me3, on the other hand, serves as an active mark for gene transcription. Recent studies show that these marks coexist and are silent in embryonic stem (ES) cells and activated during differentiation.10,11
Similar to DNA methylation, perturbation of acetylation and methylation of histone lysine residues has been observed in cancerous cells. Different types of cancers exhibit distinct histone modification patterns. In many human cancers, hyper-trimethylation of H3K27 due to elevated levels of EZH2 results in the transcriptional repression of tumor suppressor genes, leading to increased tumor cell proliferation.12-16 Therefore, normalizing H3K27me3 by targeted inhibition of EZH2 presents a potential novel method for cancer therapy. For epigenetic targets, recombinant target-based approaches are often used to identify specific small-molecule inhibitors. Although a biochemical assay is typically used to run the primary high-throughput screening (HTS), a cell-based mechanistic assay is required to measure cellular potency of compound and provide adequate support for ongoing chemistry in an active structure–activity relation (SAR) program. Examples where this approach has been deployed include identification of G9a inhibitors 17 and protein arginine N-methyltransferase (PRMT) inhibitors. 18 Typically, a Western blot using a marker-specific antibody is used to detect the ability of the compound to inhibit the histone modification. However, this method is limited for compound screening because of its very low throughput. Sandwich immunoassays such as enzyme-linked immunosorbent assay (ELISA) and dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) often represent alternative approaches to detect these epigenetic modifications because these methods are quantitative and higher throughput than Western blots. DELFIA uses lanthanide chelates in combination with time-resolved fluorometric detection and has advantages over ELISA formats in terms of sensitivity. Typically, the analyte (protein of interest) is captured by antibody immobilized on the assay plate. After a wash step, a lanthanide-conjugated antibody specific to the analyte is added and allowed to bind, followed by additional washes. The fluorescence signal can be detected by adding Enhancement Solution or DELFIA Inducer (http://www.perkinelmer.com/CMSResources/Images/44-73257FLY_DELFIAApplicationsOfTRF.pdf).
We describe herein a modified DELFIA method to quantify the H3K27me3 level in the prostate cancer cell line, PC3. Using a 384-well high-binding MaxiSorp plate and 0.2 N HCl as a lysis buffer, which is commonly used in histone extraction, 19 we were able to show that (1) histones can be captured efficiently on the plate, (2) 0.2 N HCl is more efficient than commercial cell lysis buffers in extracting H3K27me3, and (3) the assay has excellent robustness features, including a high signal-to-background ratio, low variation, and broad dynamic detection range. To exclude potential compound toxicity (i.e., cell death) on the change of methylation level of histone H3, the H3K27me3 signal was normalized to the total histone H3 in each sample well. This assay was further validated using a few known methyltransferase inhibitors. In summary, this assay, developed in a 384-well format, is sensitive, robust, and cost-effective in measuring the H3K27me3 level and suitable for either SAR support or HTS.
Materials and Methods
Materials
3-Deazaneplanocin A (DZNep) was purchased from Cayman Chemical (Ann Arbor, MI). Trichostatin A (TSA) and 12 N hydrochloric acid (HCl) were purchased from Sigma Aldrich (St. Louis, MO). SuperBlock Blocking buffer in Tris-buffered saline (TBS) and SuperBlock T20 Blocking buffer (TBS + Tween 20 [TBS-T]) were purchased from Thermo Scientific (Rockford, IL). Histone extraction buffer, 5× neutralization buffer, purified recombinant histone H3 trimethyl Lys27 (H3K27me3), histone H3 monomethyl Lys27 (H3K27me1), histone H3 acetyl Lys23 (H3K23ac), histone H3 phospho Ser10 (H3S10ph), and histone H3 proteins were purchased from Active Motif (Carlsbad, CA). DELFIA Enhancement Solution and Eu-N1-anti-rabbit IgG antibody were from PerkinElmer (Waltham, MA). Normal rabbit IgG was from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-H3K27me3 antibody was from Upstate, Millipore (Temecula, CA). Rabbit anti–histone H3 antibody was from Cell Signaling Technology (Danvers, MA). Mouse anti–histone H3 monoclonal antibody was purchased from Biosource, Invitrogen (Camarillo, CA). Fetal bovine serum (FBS) was purchased from Gibco, Invitrogen (Grand Island, NY).
Cell culture and treatment
PC3 cells, a nonrecombinant human prostate carcinoma cell line, were maintained in RPMI-1640 medium supplemented with 10% FBS. At around 80% confluency, cells were trypsinized, washed, and resuspended in RPMI-1640 medium with a specified FBS concentration. Cells were added to a 384-well clear-bottom cell culture plate from Greiner Bio-One (cat. 781091; Monroe, NC) at 50 µL/well volume at desired cell densities. Cells were incubated overnight at 37 °C with 5% CO2 and then treated with DMSO or compounds for different time periods before DELFIA detection of H3K27me3.
Cell lysis
In this report, several approaches were used to prepare cell lysate and extract histone proteins. For cell culture plates, the medium was first aspirated and the plates were frozen at −80 °C for at least 1 h (typically, plates were stored at −80 °C until use). Prior to assay, the frozen plates were equilibrated at room temperature for 30 min. As defined in the figure legends, cells were either lysed with detergent lysis buffer (25 mM Tris-HCl [pH 7.4], 250 mM NaCl, 2 mM MgCl2, 1 mM CaCl2, 1% NP-40, 1% Triton X-100, 1% NaDeoxycholate, 1 mM PMSF, 1× protease inhibitor cocktail, and 20 U/mL DNase I), a commercial histone extraction buffer, or 0.2 N HCl for a specific time period at either room temperature or 4 °C. After lysis, the cell mixture was pipetted up and down for 10 cycles with a CyBi-Well pipettor (CyBio, Boston, MA). The plates were then spun for 5 min at 800 rpm at room temperature in a bench-top plate centrifuge. The cell lysate was finally transferred with a Cybi-Well to a Nunc high-binding MaxiSorp plate (eBioscience, San Diego, CA) for detection as described below.
Compound handling and tool compound testing
Compounds were dissolved in neat DMSO to 10 mM as stock solutions. From the 10-mM stock solution, compounds were diluted to desired concentrations in a 384-well master plate. An Echo liquid handling system (Labcyte, Sunnyvale, CA) was used to stamp the compounds from the master plate into the cell plates. Cells in column 6 were treated with DMSO (1% final) and detected with the specific anti-H3K27me3 antibody as the high control (no decrease of H3K27me3, 0% response). Cells in column 18 were also treated with DMSO (1% final) but detected with normal rabbit IgG as the low control (100% decrease of H3K27me3, 100% response). To perform compound dose–response curves, PC3 cells were resuspended in RPMI-1640 medium supplemented with 5% FBS, plated at 2000 cells/well, and incubated overnight. DZNep or TSA was then added to the plates at different doses and incubated for an additional 2 or 4 days.
DELFIA detection of H3K27me3 and total histone H3
A DELFIA method was employed to measure the level of H3K27me3 and total histone H3. Compound-treated cells (see above) were lysed with 50 µL/well of 0.2N HCl. Then, 20 µL of cell lysate was transferred to two 384-well high-binding MaxiSorp plates. One copy was used for detection of H3K27me3; the other was used for detection of total histone H3. After cell lysate transfer, 5 µL/well of 5× neutralization buffer was added. The plates were then incubated for 1 h at room temperature, followed by one wash with TBS-T at 50 µL/well and two washes with TBS-T at 100 µL/well. All the wash steps were performed with a BioTek EL406 plate washer (BioTek, Winooski, VT). For blocking, 100 µL/well of SuperBlock blocking buffer was added and incubated for at least 1 h at room temperature. The plates were washed three times with 100 µL/well of TBS-T washing buffer. The detection antibody (rabbit anti-H3K27me3 or rabbit anti–histone H3 antibody) was then added at 25 µL/well to the whole plate except column 18. The rabbit anti-H3K27me3 antibody was diluted in SuperBlock-T20 to 1 µg/mL. The rabbit anti–total histone H3 antibody was diluted in SuperBlock-T20 to 0.29 µg/mL. After a 1-h incubation at room temperature, the plates were washed three times at 100 µL/well of TBS-T. Then, 25 µL/well Eu-N1-labeled anti-rabbit IgG antibody, which was diluted 3000-fold in SuperBlock-T20, was added. After a 1-h incubation, the plates were washed three times with 100 µL/well of TBS-T and two times with 120 µL/well of TBS-T. Next, 25 µL/well of DELFIA Enhancement solution was added. After a 30-min incubation at room temperature, the plates were read with an EnVision plate reader (PerkinElmer, Waltham, MA) with excitation wavelength at 320 nm, emission wavelength at 615 nm, and a 505-nm dichroic mirror. For DELFIA experiments performed with purified recombinant proteins, these proteins were first diluted to desired concentrations and then added directly to a MaxiSorp plate. The same procedures were then followed for DELFIA detection.
Data processing and analysis
Total histone H3 was measured in a similar manner using anti-H3 antibody and was used to normalize the signal of H3K27me3 in each sample well to mitigate cell viability changes after compound treatment. The normalization for H3K27me3 signal was calculated as follows:

The normalized signal of H3K27me3 was then used for pIC50 calculation of compounds using GraphPad Prism (GraphPad Software, La Jolla, CA).
Results and Discussion
Capturing of histone H3 proteins
We initially coated anti–histone H3 antibody on the plate to capture histone H3 for further detection. In these experiments, MaxiSorp plates were coated with different amount of mouse anti–histone H3 mAb. Various concentrations of purified recombinant H3K27me3 were then added to the antibody-coated plates followed by DELFIA detection. As shown in Figure 1A , the DELFIA signal increased with added H3K27me3 in a dose-dependent manner. Interestingly, it was observed that the signal was virtually independent of the amount of capture antibody in the well. In fact, in the absence of mouse anti–histone H3 antibody, the signal was essentially the same or even slightly higher than in the presence of capture antibody. This result suggested that histone H3 protein was able to bind directly to the MaxiSorp plate in the absence of capture antibody. To test this hypothesis, we directly added recombinant H3K27me3, H3K27me1, or histone H3 proteins in a 384-well MaxiSorp plate and then performed the DELFIA assay using anti-H3K27me3 Ab as the detection antibody. As shown in Figure 1B , DELFIA signal in H3K27me3-coated wells showed a dose-dependent increase, whereas the signal in H3K27me1- and histone H3–coated wells remained at a basal level under all tested doses. When an anti–histone H3 was used as the detection antibody, these three recombinant proteins all gave a dose-dependent DELFIA signal (data not shown). These results confirmed that a capture antibody for histone H3 was not needed and that the three recombinant proteins were able to bind directly to the MaxiSorp plate. In addition, detection using anti-H3K27me3 antibody generated a specific signal only in the presence of H3K27me3.
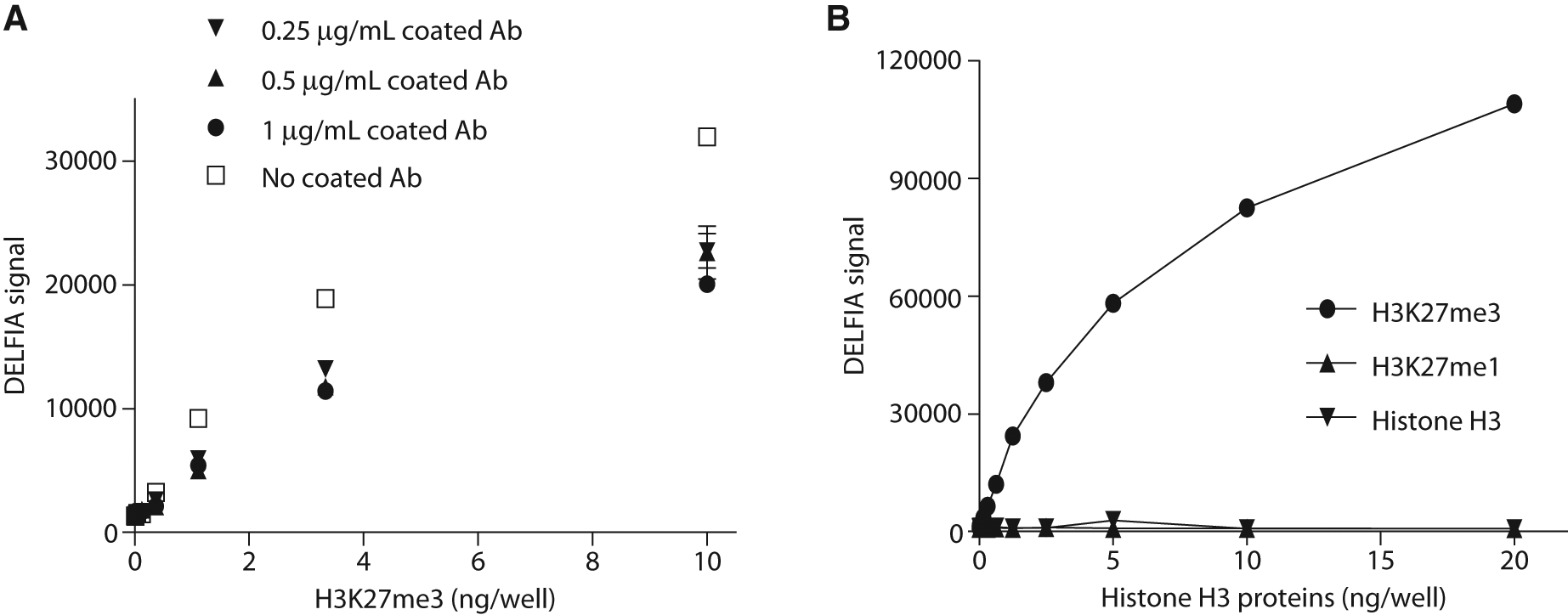
Plate-based capture of histone H3 proteins and detection of H3K27me3. (
After these experiments, we tested different coating conditions. These included testing different coating buffers, incubation times, temperature, and an evaluation of another source of anti–histone H3 antibody. All these different conditions revealed a consistent result that the DELFIA signal for H3K27me3 was not affected by different antibody amounts or coating conditions, thus confirming that coating the plate with anti–histone H3 antibody was unnecessary for the immobilization of H3K27me3 to the MaxiSorp plate.
Similarly, in an assay to identify histone acetyltransferase inhibitors, Turlais et al. 20 reported that histone H3 protein was able to bind directly to scintillation microplates. In a separate report, Kamei et al. 21 used calf thymus histone H2B to coat wells of MaxiSorp plates for ELISA detection of anti–histone H2B antibody levels in sera. Consistent with these reports, our data demonstrated that histone H3 proteins bound directly to MaxiSorp plates. The lack of a requirement for a capture antibody for immobilization of histone proteins simplifies the assay logistics. However, one should be cautious when mixing a non–histone protein target with histone proteins as the presence of histone proteins could potentially interfere with the binding to the plates of other target proteins.
Binding affinity and detection of H3K27me3 from the pooled fraction and cell lysate
The above data demonstrated that recombinant H3K27me3 was able to bind to the MaxiSorp plate, and it can be detected quantitatively in the absence of a capture antibody. We then tested if the presence of other modified H3 protein or cell lysate would affect the binding and detection of recombinant H3K27me3. We compared the binding and detection of H3K27me3 alone and in the presence of other types of modified histone H3 (all recombinant material) as well as in the presence of PC3 cell lysate. H3K27me3 was mixed and serial-diluted with an equal amount of one other type of modified histone H3 or a pooled fraction and was added to a MaxiSorp plate at varying concentrations. As shown in Figure 2A , detection of H3K27me3 did not change significantly when the protein was plated in the presence of histone H3, H3K27me1, H3S10ph, or H3K23ac. There was a lower signal when H3K27me3 was mixed and plated in the presence of the pooled fraction consisting of four other histone H3 proteins. This was likely due to the overwhelming presence of other H3 proteins in competing with H3K27me3 binding to the plate and the binding capacity of the plate being exceeded. This experiment demonstrated that different modifications of histone proteins did not significantly affect the binding affinity of H3K27me3 to a MaxiSorp plate. Similarly, we also mixed H3K27me3 with PC3 cell lysate and applied this to a MaxiSorp plate for detection. To carry out this experiment, PC3 cells were cultured overnight and plated in a 384-well cell culture plate at 16K cells/well and then lysed in the plate by addition of 50 µL/well of detergent lysis buffer. The plate was incubated for 30 min at room temperature with agitation. After which, the plate was centrifuged, and 20 µL/well of the supernatant was transferred to a MaxiSorp plate. Immediately following this step, 5 µL of recombinant H3K27me3 was added to the plate. In parallel, recombinant H3K27me3 was added to wells with detergent lysis buffer instead of cell lysate. As shown in Figure 2B , in the presence of PC3 cell lysate, the DELFIA signal increased in a dose-dependent manner to the added H3K27me3. In comparison, the signal in the presence of PC3 cell lysate was substantially lower than that of H3K27me3 alone, but more important, the signal was still dose dependent. This was likely because other proteins in the cell lysate, including histones, competed with the binding of recombinant H3K27me3 to the plate as described earlier.
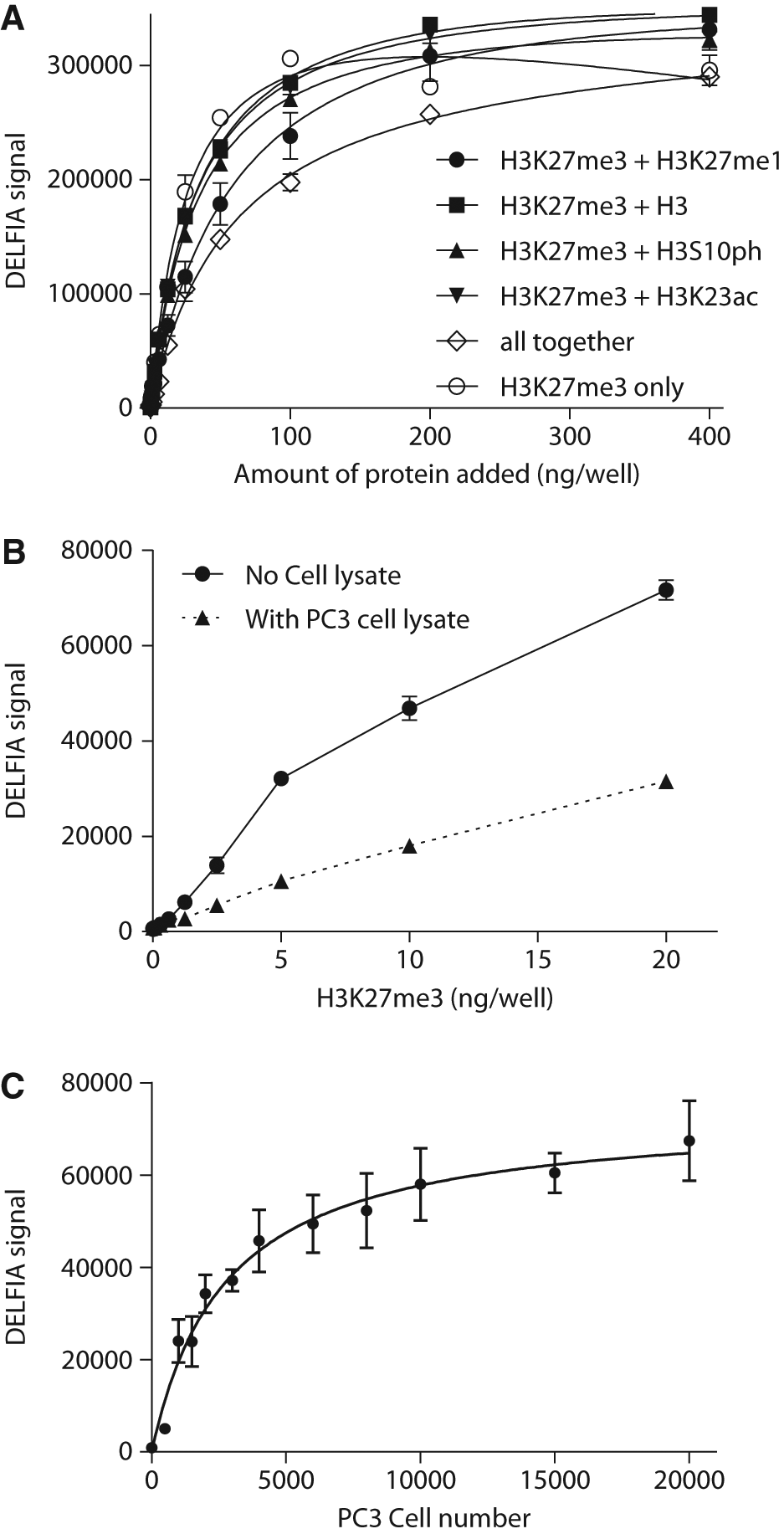
Dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA)–based detection of H3K27me3. (
The above data showed that the binding affinity of H3K27me3 to the MaxiSorp plate was not perturbed in the presence of other types of histone H3 proteins. In addition, the dose-dependent nature of the assay was maintained, albeit at a lower signal, in the presence of the PC3 cell lysate. To maximize the sensitivity of the assay in detecting the change of H3K27me3 level, we next analyzed the dynamic detection range of H3K27me3 from PC3 cell lysate. PC3 cells were grown in large flasks, trypsinized, washed, and then counted. Different numbers of cells were pelleted in 1.5 mL microfuge tubes and lysed by repeated pipetting in 40 µL of 0.2 N HCl. The lysates were then incubated at 4 °C overnight and centrifuged for 10 min at 4 °C in a microcentrifuge. The supernatants were transferred to a MaxiSorp plate at 25 µL/well for detection of H3K27me3 levels by DELFIA. As shown in Figure 2C , the signal increased in a cell number–dependent manner with the saturation point approximately 20K cells/well. The raw counts of DELFIA signal had a very robust readout to above 50 000, whereas the basal readout was only a few hundred. The goal was to measure the decrease in H3K27me3 level after compound treatment. We hence selected assay conditions that yielded a starting signal (in the absence of compound treatment) in the relatively high range to achieve a good assay window. Based on the curve of Figure 2C , adding around 4000 to 5000 cells per well yielded a relatively high DELFIA signal but still within the linear range of detection.
Noticeably, under similar cell density, the cell lysate in Figure 2C had a much higher signal compared with that in Figure 2B . This difference strongly suggested that the cell lysate prepared in the 384-well plate with the detergent buffer ( Fig. 2B ) had a lower concentration of H3K27me3 than that prepared from cells lysed with 0.2 N HCl in microfuge tubes ( Fig. 2C ). Cell lysate prepared in the microfuge tube with multiple cycles of pipetting clearly showed that a significant amount of histone proteins was extracted. This is likely due to the higher efficiency of lysis of resuspended cells in the microfuge tubes than when cells were lysed directly in the assay plate. Because the compounds were to be tested in 384-well plates, we next set out to find a method that could sufficiently and homogeneously extract histone proteins directly from cells cultured in 384-well plates.
Lysis of cells and extraction of histone proteins in 384-well plates
We plated PC3 cells in a 384-well clear-bottom cell culture plate and incubated the plates at 37 °C overnight and then tested different methods for the extraction. We first used detergent lysis buffer to prepare cell lysate by incubating at room temperature for 30 min or overnight at 4 °C with gentle shaking. Using this method, the signal/background (S/B) ratio was only around 4 for plates that had been subjected to a 30-min incubation at room temperature. In comparison, overnight incubation at 4 °C totally diminished the S/B ratio ( Table 1 ). This was possibly due to protein degradation during the long incubation time, even when the lysis was performed in the presence of protease inhibitors. Sonication was also tried to increase the efficiency of histone extraction. Although it did boost the S/B ratio to around 13, there was a very high variation among samples (data not shown). This was likely due to uneven sonication of the plate in a water bath sonicator. In addition, the sonciation was not applicable for HTS and was therefore not explored further.
Assay Signal-to-Background (S/B) Ratio and Z′ under Various Lysis Conditions

PC3 cells were plated into a 384-well cell culture plate at 10 000 cells/50 µL/well. Cells were incubated at 37 °C, 5% CO2 overnight. After removing the cell culture medium, different lysis buffer was added at 50 µL/well and incubated as indicated. Then, 20 µL/well was transferred to a 384-well MaxiSorp plate for detection of H3K27me3.
We next tested a commercial histone extraction buffer. Although the robust Z′ was significantly improved to 0.49 ( Table 1 ) as compared with the Z′ obtained by the use of detergent buffer, the S/B ratio was only slightly increased to 5.7. The low S/B indicated that the histone extraction was still not complete. We then tried the traditional acid extraction of histone proteins with 0.2 N HCl. 19 We added 50 µL/well of 0.2 N HCl in each well and incubated at 4 °C overnight to lyse the cells. Using this method, the S/B ratio was dramatically increased to more than 100 with robust Z′ above 0.8. These data demonstrated that incubation with 0.2 N HCl efficiently extracted histone proteins from cells cultured in 384-well plates. These data also demonstrated that histone protein degradation was not an issue with overnight incubation in the presence of 0.2 N HCl. We therefore adopted this plate-based lysis method for further assay optimization. During methods development, we also found that freezing the cell plates before lysis resulted in slightly higher S/B ratios. If needed, the lysis step can also be completed in an hour at room temperature, instead of overnight at 4 °C (data not shown).
Compound treatment of PC3 cells and normalization of H3K27me3 signal
As described above, a robust 384-well plate-based H3K27me3 detection assay was established using cells cultured at 10 000 cells per well followed by overnight incubation. We next optimized the compound treatment conditions, including cell density, treatment time, and serum concentrations. As shown in Figure 2C , 4000 cells per well had relatively high signal but still in the linear range of detection. Because the lysate in Figure 2C was prepared in an Eppendorf tube, which was more efficient for histone H3 extraction than in a 384-well plate, we asked whether we could further decrease cell density to improve assay sensitivity. To this end, we evaluated 2000 cells/well and 4000 cells/well and found that 2,000 cells/well gave an S/B ratio of 30 and Z′ of 0.7 (data not shown), which is an assay signal that is sufficiently robust for an HTS assay.
We then optimized the treatment time and the assay serum concentration in the presence of an inhibitor. We initially used TSA to treat PC3 cells in an attempt to decrease the amount of H3K27me3 through the inhibition of histone deacetylation. However, the data suggested that most of doses of TSA were cytotoxic and did not cause the levels of H3K27me3 to decrease ( Fig. 3B ). We then used another tool compound 3-deazaneplanocin A (DZNep) to optimize the assay conditions. DZNep is a global histone methylation inhibitor. 22 As shown in Figure 3A , treatment of PC3 cells with DZNep resulted in a dose-dependent H3K27me3 signal decrease under all the conditions tested, and a four-day treatment had a more profound decrease of H3K27me3 signal than a two-day treatment. This indicated that change of H3K27me3 level in the PC3 cells required a relatively long exposure time to the compound. This is perhaps due to the nature of epigenetic modifications as cell replication is required for these modifications to take effect. Although long-term treatment of cells produced better response to the test compounds, it also increases the chance of the cytotoxicity. This is indeed a common concern for cell-based assays. In fact, during the assay development, we observed significant cytotoxicity of several tested tool compounds such as TSA and SAHA (data not shown). It is therefore critical to measure the total histone H3 level and compare the response of compound while taking into account cell health to accurately monitor the change of methylation of H3K27.
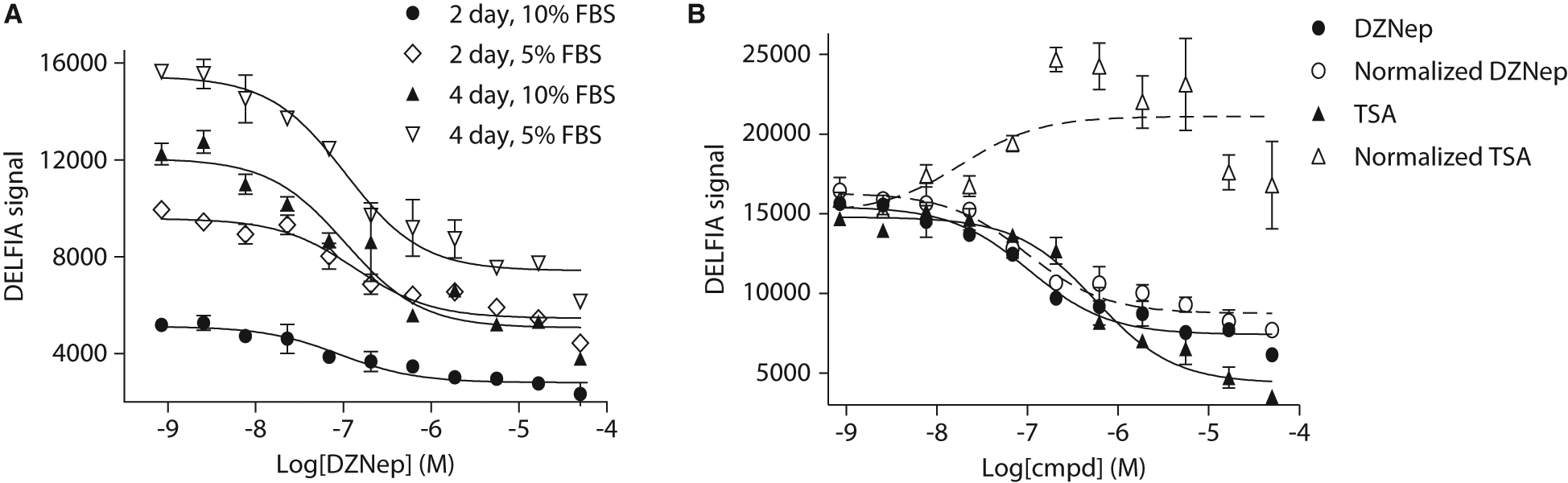
(
Toward this goal, we measured total histone H3 in parallel and used it to normalize the level of H3K27me3. Equal volume of cell lysate from compound-treated cells was transferred to two separate MaxiSorp plates. One plate was processed for detection of H3K27me3, whereas the other plate was for detection of total histone H3. If a sample well has the same level of total histone H3 as the DMSO treated well, the fold change of total histone H3 would be 1. This would suggest the treatment had no significant effect on cell viability. Accordingly, the normalized H3K27me3 signal would remain the same as the measured signal. If the total histone H3 level in a sample well is only half of that in a DMSO-treated well, the fold change would be 0.5. This would indicate that treatment of compound causes cell numbers to decrease. Therefore, the normalized H3K27me3 signal would be doubled. Figure 3B compared the measured H3K27me3 signal with the normalized H3K27me3 signal of compound-treated samples. DZNep treatment generated a dose-dependent signal decrease with a pIC50 value around 7.0 ( Fig. 3B ). In comparison, the normalized H3K27me3 signal ( Fig. 3B ) was only slightly higher than the measured H3K27me3 signal, suggesting a very mild cytotoxicity after the four-day treatment. The pIC50 values generated from these two dose–response curves were essentially the same. In contrast, even though the TSA treatment showed a dose-dependent decrease of the H3K27me3 signal, the curve generated using normalized data showed a dose-dependent increase, albeit the curve was shallow ( Fig. 3B ). This can be explained by the following reasons. First, TSA treatment does not affect H3K27me3 level. Second, TSA treatment causes total histone H3 level to decrease because of its toxicity. In fact, we observed that TSA caused cell viability to decrease at around or above 100 nM after overnight treatment in A549 and HEK293 MSRII cells using the Cell Titer Glo assay (data not shown). Taken together, these data clearly demonstrate that using total histone H3 to normalize the H3K27me3 signal effectively excluded interference from cell viability caused by compound treatment. This normalization was easy to perform and effective to reduce false positives. In addition, changes in total histone H3 level alone could be used as an indicator of compound cytotoxicity.
It is noticeable that the assay widow of DZNep was relatively shallow, although a clear dose-dependent decrease of H3K27me3 was evident. This was in agreement with data generated from Hayden et al., 23 who also reported a shallow dose–response curve in a cell proliferation assay after cells were treated with DZNep. Meanwhile, several in-house methyltransferase inhibitors were also tested in this assay. Data revealed a much higher assay window (~6-fold vs. 2.5-fold for DZNep) for both measured and normalized H3K27me3 curves (data not shown). This line of evidence suggests that the assay has a good dynamic range and is suitable for screening and SAR support.
In summary, the DELFIA method we described herein is able to efficiently and robustly measure the change of H3K27me3 level in PC3 cells treated with methyltransferase inhibitors in a 384-well plate. It should be noted that this assay monitors the change of endogenous histone proteins in PC3 cells, and no overexpression of recombinant protein is required. By normalizing the H3K27me3 signal with total histone H3 signal in each sample well, this assay produces a readout that is target specific rather than detecting cytotoxicity. Finally, the method established in this report for H3K27me3 can be applied to the development of high-throughput plate-based assays for other histone proteins.
Footnotes
Acknowledgements
We thank Caretha L. Creasy in the Cancer Epigenetics Discovery Performance Unit, Cancer Research for her critical reading of the manuscript; Sample Management Technology Group for their help with compound management; and Gordon McIntyre and Thomas Meek for their support in carrying out this work.