
Abstract
The nicotinic acetylcholine receptors (nAChRs) are a member of the ligand-gated ion channel family and play a key role in the transfer of information across neurological networks. The X-ray crystal structure of agonist-bound α7 acetylcholine binding protein (AChBP) has been recognized as the most appropriate template to model the ligand-binding domain of nAChR for studying the molecular mechanism of the receptor–ligand interactions. Virtual screening of the National Cancer Institute diversity set, a library of 1990 compounds with nonredundant pharmacophore profiles, using AutoDock against AChBPs revealed 51 potential candidates. In vitro radioligand competition assays using [3H] epibatidine against the AChBPs from the freshwater snails, Lymnaea stagnalis, and from the marine species, Aplysia californica and the mutant (AcY55W), revealed seven compounds from the list of candidates that had micromolar to nanomolar affinities for the AChBPs. Further investigation on α7nAChR expressing in Xenopus oocytes and on the recombinant receptors with fluorescence resonance energy transfer (FRET)–based calcium sensor expressing in HEK cells showed that seven compounds were antagonists of α7nAChR, only one compound (NSC34352) demonstrated partial agonistic effect at low dose (10 µM), and two compounds (NSC36369 and NSC34352) were selective antagonists on α7nAchR with moderate potency. These hits serve as novel templates/scaffolds for development of more potent and specific in the AChR systems.
Introduction
Nicotinic acetylcholine receptors (nAChRs) are important members of the pentameric ligand-gated ion channel superfamily that are composed of five homologous subunits organized around a central pore. 1 nAChRs are divided into two groups: (1) the muscle type, found in vertebrate skeletal muscles, where they mediate neuromuscular transmission at the neuromuscular junction, and (b) the neuronal type, found mainly throughout the peripheral nervous system and central nervous system (CNS) but also in nonneuronal tissues (keratinocytes, epithelia, macrophages, etc.). 2 In the CNS, neuronal nAChR subtypes are involved in a number of processes connected to cognitive functions, learning and memory, arousal, reward, motor control, and analgesia.3,4 Abnormal opening and closing of nAChRs contributes to neurodegenerative disorders, resulting in severe diseases such as Alzheimer disease, Parkinson disease, dyskinesias, Tourette syndrome, and schizophrenia. 3 Among all the subtypes identified to date, α7 and α4β2 have been established as two major targets mediating the pathology of these severe diseases.5–7 Compared with other nAChRs, the α7 receptor mediates the neuron-protective action of nicotine-like agonists against various stresses, including β-amyloid and nerve growth factor depletion.8–10 Selective α7 agonists can prevent receptor activation by β-amyloid and do not possess significant drug dependence, making the α7 agonist a potential candidate for the treatment of Alzheimer disease.8,11,12 In addition, several studies have identified links between nonneuronal α7nAChR subtypes and a number of conditions and diseases, including inflammation and cancer.13–15 α7nAChRs are expressed on bronchial epithelial and non–small cell lung cancer cells (NSCLC) and are involved in cell growth regulation.16,17 High-affinity α7nAChR inhibitors can induce apoptosis in NSCLC and malignant pleural mesothelioma (MPM) and inhibit angiogenesis.15,18,19 Exploitation of such pharmacologic properties can lead to the discovery of new specific cholinergic antagonists as anticancer therapies.
Contemporary understanding of the structure–function relationship of nAChRs has greatly benefited from the two major breakthroughs achieved so far: the refined structure of Torpedo α 1 β1δγ nAChR and several crystal structures of the acetylcholine binding protein (AChBP) in different agonist/antagonist-bound states.20–23 Although the refined structure of α1β1δγ nAChR at 4 Å resolution has provided fundamental insights into the architecture of the extracellular ligand binding domain (LBD) and the channel pore, the binding site is obviously distorted, making it impossible to offer in-depth information about ligand binding. 20 Overall, AChBP shares about 24% sequence identity with LBD of nAChR and has the same pentameric assembly. 21 The X-ray crystal structure of agonist-bound AChBP has been recognized as the most appropriate template to model the LBD of nAChR for the purpose of studying the molecular mechanism of receptor–ligand interactions.22–25 The AChBP molecule has been identified from the freshwater snail, Lymnaea stagnalis (Ls)26,27; the marines species, Aplysia californica (Ac) 28 ; and the freshwater species, Bulinus truncatus (Bt). 29
Virtual screening has been widely used to discover new lead compounds for drug design. 30 Successful studies have resulted in the discovery of molecules resembling either the native ligands of the specific targets or novel leads. Therefore, this study aimed to find novel agonists or antagonists that could more potently and selectively interact with AChR.
Materials and Methods
Materials
Binding protein was prepared at Taylor’s lab, Skaggs School of Pharmacy and Pharmaceutical Sciences at the University of California, San Diego. Anti-mouse SPA was purchased from Amersham Biosciences (Piscataway, NJ). All other chemicals were of reagent grade and purchased from Sigma (St. Louis, MO). The tested 51 compounds from virtual screening were from the National Cancer Institute (NCI; http://dtp.nci.nih.gov) provided by Olson’s lab, The Scripps Research Institute (San Diego, CA). The cDNA clones of human α7nACh were kindly provided by Dr. H. Tsumeki (Faculty of Pharmaceutical Sciences, University of Toyama, Japan). The circular cDNAs of α7nACh were linearized with NOT I. The linearized cDNAs were then transcripted into cRNAs in vitro with T7 RNA polymerase, using a mMessage Machine transcription kit (Ambion, Austin, TX). The cRNAs were diluted with nuclease-free water to approximately 0.5 µg/µL.
Target preparation
Acetylcholine binding protein complex structures with agonist ligand were found in the Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do). 1UW6, 22 2BYQ, 23 and 1BJ0 29 were the crystal structures of the pentameric AChBP from Ls, Ac, and Bt, respectively. The ligand from each AChBP complex was selected for the validation study. Polar hydrogen atoms were added and Gasteiger charges were assigned to all atoms. The grid maps representing the ligand were calculated with AutoGrid. The dimensions of the grid were 40 × 40 × 40 grid points with a spacing of 0.375 Å between the grid points and centered on the ligand. Each ligand was redocked to validate the prepared nAChBP active binding sites.
NCI diversity set
The NCI diversity set is a reduced set of 1990 compounds selected from the original NCI-3D structural database for their unique pharmacophores (http://dtp.nci.nih.gov/branches/dscb/diversity_explanation.html). All hydrogens were added, Gasteiger charges were assigned, 31 and then the nonpolar hydrogens were merged. The rotatable bonds were assigned via AutoTors. 32
Molecular docking
The program AutoDock3.0.5 32 from The Scripps Research Institute uses a Larmarckian genetic algorithm for docking flexible ligands into protein binding sites to explore the full range of ligand conformational flexibility with the rigid protein. The AutoDock run parameters used were as follows: for each compound in the library, the number of genetic algorithm (GA) runs was 100, the population size was set to 150, and the maximum number of energy evaluations was increased to 10 000 000 per run. All other run parameters were kept at their default settings. The jobs were run on Bluefish, a 64-bit 576-processor LINUX cluster at The Scripps Research Institute. Final docked conformations were clustered using a tolerance of 2 Å root mean square deviation (RMSD). The lowest docking energy of the 100 dockings for each AChBP (1UW6, 2BYQ, and 2BJ0) was ranked. The top 51 compounds that matched all three AChBPs with the lowest docking energies, a ligand efficiency (LE) lower than −0.30, and “drug-like” properties were selected for experiment testing.33,34
Effect of hits on the binding of AChBP
An adaptation of a scintillation proximity assay (SPA) was employed to determine the apparent Kd value. 35 AChBP (final concentration ~500 pM binding sites for Ls and Bt, ~1 nM binding sites for Ac), polyvinyltoluene anti-mouse SPA scintillation beads (0.1 mg/mL), monoclonal anti-FLAG M2 antibody from mouse, and [3H] epibatidine (5 nM final concentration for Ls and AcY55W, 20 nM for Ac) were combined in a phosphate buffer (0.1 M, pH 7.0) with fixed concentrations of the competing ligands at 10 µM in a final volume of 48 µL. Total binding was determined in the absence of the competing ligand, and nonspecific binding was measured by adding a saturating concentration (15 µM) of the competitive ligand methyllycaconitine (MLA). The resulting mixtures were allowed to equilibrate at room temperature for a minimum of 2 h and measured on an LS 6500 liquid scintillation counter. The result was calculated by the fraction of [3H] epibatidine. The ligand with a fraction of [3H] epibatidine lower than 0.50 was selected for measuring its Kd values via [3H] epibatidine with increasing concentrations of the competing ligand in a final volume of 48 µL. The data obtained were normalized and fit to a sigmoidal dose–response curve (variable slope), and the Kd was calculated from the observed EC50 value using GraphPad Prism version 4.02 for Windows (GraphPad Software, San Diego, CA).
Functional assay using α7nAChR expressed in Xenopus oocytes (oocyte injection)
Oocyte injection
Xenopus laevis (Xenopus Yoshyoku-kyouzai, Ibaraki, Japan) were anesthetized in ice water, and a lobe of the ovary was dissected and placed in sterile modified Barth’s solution (MBS: 88 mM NaCl, 1 mM KCl, 0.41 mM CaCl2, 0.33 mM Ca(NO3)2, 0.82 mM MgSO4, 2.4 mM NaHCO3, 7.5 mM Tris-(hydroxymethyl)amino-methane, pH 7.6). Oocytes were then isolated manually and defolliculated by incubation in 1.5 mg/mL collagenase (type IA; Sigma) at 19 °C for 1 h in calcium-free MBS solution. The cRNA (50.0 nL) encoding α7nAChR was injected into oocytes, stage V to VI, with a microinjector (Drummond, Broomall, PA). For expression, the oocytes were incubated in MBS containing 2.5 U/mL penicillin and 2.5 µg/mL streptomycin at 18 °C for 2 to 3 days before recording.
Voltage-clamp recording in X
laevis oocytes. Responses to acetylcholine (ACh) were recorded with a two-electrode voltage-clamp amplifier (GeneClamp 500B; Axon Instrument, Foster City, CA) at a holding potential of −80 mV. Electrodes were filled with 3 M KCl and had resistances of 1 to 5 MΩ. Oocytes injected with cRNA encoding α7nAChR were placed in a 50-µL chamber and continuously perfused with low-calcium Ringer’s solution (82.5 mM NaCl, 2.5 mM KCl, 0.5 mM CaCl2, 2 mM MgCl2, 5 mM Tris-(hydroxymethyl) amino-methane, pH 7.6) at 1 mL/min at room temperature. To examine the antagonistic action of the compound on α7nAChR, each oocyte received initial control applications of ACh, applications of hit compounds, and then a follow-up control application of ACh. Drugs were diluted in perfusion solution and applied with a solenoid valve to switch from perfusion to drug solutions. Each oocyte was tested for positive expression by performing a test perfusion with 100 µM ACh. Oocytes with resting membrane potentials between −20 and −40 mV and displaying inward currents of at least 20 nA in response to ACh were used in experiments. All data were taken from three to five different oocytes. In the experiment, the NCS hit compounds were dissolved in 100% DMSO; the final concentration of DMSO used in this study was less than 0.01% and had no pharmacological effect when applied alone. MLA, a toxin derived from the seeds of Delphinium brownii and reported to be an α7-selective antagonist at low concentrations, was used as a control for antagonistic effect. The results are expressed as percentages of control responses to control for the variability in the number of receptors expressed in different oocytes. The control responses were measured before and after drug application. All values are presented as mean ± SEM.
Functional assay using recombinant receptors and FRET-based calcium sensor expressing in HEK cells
Further functional assay for selectivity was performed using sensor cells expressing Ca2+-permeable ligand-gated ion channel (LGIC) receptors and a genetically encoded fluorescence resonance energy transfer (FRET)–based calcium sensor (or cell-based neurotransmitter fluorescent engineered reporters, CNiFERs). 36 HEK cells expressing α7nAChR CNiFERs and 5-HT3A CNiFERs were analyzed by FRET response employing a fluorometric imaging plate reader system (FlexStation 3; Molecular Devices, Sunnyvale, CA), whereas the cells expressing α4β2 and α1nAChR CNiFERs were tested in the FlexStation 3 system with a membrane potential–sensitive dye. The cells were plated in 96-well clear-bottom, poly-D-lysine-coated black microplates (Costar; Corning, Corning, NY) for 24 h prior to experiments.
For testing of FRET responses for α7nACh and 5-HT3A receptors, the medium was then replaced with 100 µL of artificial cerebrospinal fluid (aCSF, containing 121 mM NaCl, 5 mM KCl, 26 mM NaHCO3, 12 mM NaH2PO4, 10 mM glucose, 1.2 mM CaCl2, 1.3 mM MgSO4, 5 mM HEPES Na+, pH 7.4) for 5-HT3A CNiFERs and aCSF with 15 µM of PNU-120596 for α7nAChR CNiFERs. The plate was then incubated at 37 °C for 30 min. The compounds were prepared in aCSF buffer as 3× solutions in a separate 96-well polypropylene plate. Experiments were conducted at 37 °C using 436-nm excitation. Emitted light was collected at 485 nm and 528 nm. Basal fluorescence was recorded for 10 s, followed by addition of 50 µL of ligand (first addition). Measurements were made at 1.52-s intervals for 1.5 min to assess the agonistic effect of test ligand. After being incubated at 37 °C for 30 min, agonist was added to each well to assess antagonism of agonist response. Then, 100 nM epibatidine and 1 µM 5-HT as a final concentration were used as agonists for α7nACh and 5-HT3A receptors, respectively. The responses were normalized to the response of agonist, and inhibitory activity was calculated from 100 subtracted by the percentage of agonist response.
Activities of compounds at α4β2 and α7nAChRs were determined using a membrane potential–sensitive fluorescent dye. Growth media were removed from the cells, and membrane potential dye (Molecular Devices), reconstituted in two times more diluted than the manufacturer’s instructions, was added to the wells. Plates were incubated for 30 min at room temperature and then directly transferred to the FlexStation 3 system. The compounds were prepared in aCSF buffer as 3× solutions in a separate 96-well polypropylene plate. Experiments were conducted at 37 °C using 530-nm excitation. Emitted light was collected at 565 nm every 1.52 s. Baseline fluorescence was monitored for the first 10 s, followed by the addition of compounds. Then, 100 nM epibatidine and 10 µM succinylcholine as a final concentration were used as agonists for α4β2 and α1nAChRs, respectively. The agonist and antagonist activities of the compounds were assessed as described before.
Results and Discussion
Virtual screening and docking results
Molecular docking was carried out to investigate the docking energy and binding mode of compounds to AChBP using AutoDock program version 3.0.5. 32 Although AChBP shares only 24% sequence identity with the LBD of α7nAChR, the AChBP was used as a surrogate structure of this receptor because of its great secondary structural similarity to the LBD of nAChR and the high level of sequence identity at the binding site (52%). 37 In addition, there are a number of high-resolution structures in PDB of both agonist-bound AChBP complexes and antagonist-bound AChBP complexes. Among three different species of the agonist-bound AChBP—1UW6, 22 2BYQ, 23 and 2BJ0 29 —were crystal structures of nicotine, epibatidine, and 3-cyclohexyl-1-propylsulfonic acid bound to the pentameric AChBP, respectively. The AChBP structures from chains A and B from each BP were used as templates for virtual screening, and the ligand between that chain was used to validate the template of AChBP. The ligand from each crystal structure was redocked to be the control to validate the AChBP binding site. The result shows that 100% of the docked conformations grouped into a single cluster using an RMSD clustering tolerance of 2.0 Å, and the docked orientation was close to that of the crystal structure with RMSD of 1.08, 1.38, and 1.10 Å for 1UW6, 2BYQ, and 2BJ0, respectively. The redocking result indicated that the prepared AChBP templates are a good model for docking studies of the NCI diversity set of 1900 compounds.
For virtual screening, after each of 100 docking runs per compound, the conformations that had the lowest docked energy of binding to each AChBP (1UW6, 2BYQ, and 2BJ0) were clustered and ranked. Each cluster consisted of conformers that had similar 3D structures (RMSD < 2 Å). The top-ranking compounds that matched the three AChBP templates and adhered to Lipinski’s rule of five were filtered for experimental testing. The cutoff values were −10 kcal/mol for binding free energy and −0.3 for ligand efficiency. Only 48 compounds that met the selection criteria and 3 more compounds with good pharmacodynamic properties (low binding free energy and high percent membership in the largest cluster) were included (see
Seven Hit Compounds from Virtual Screening
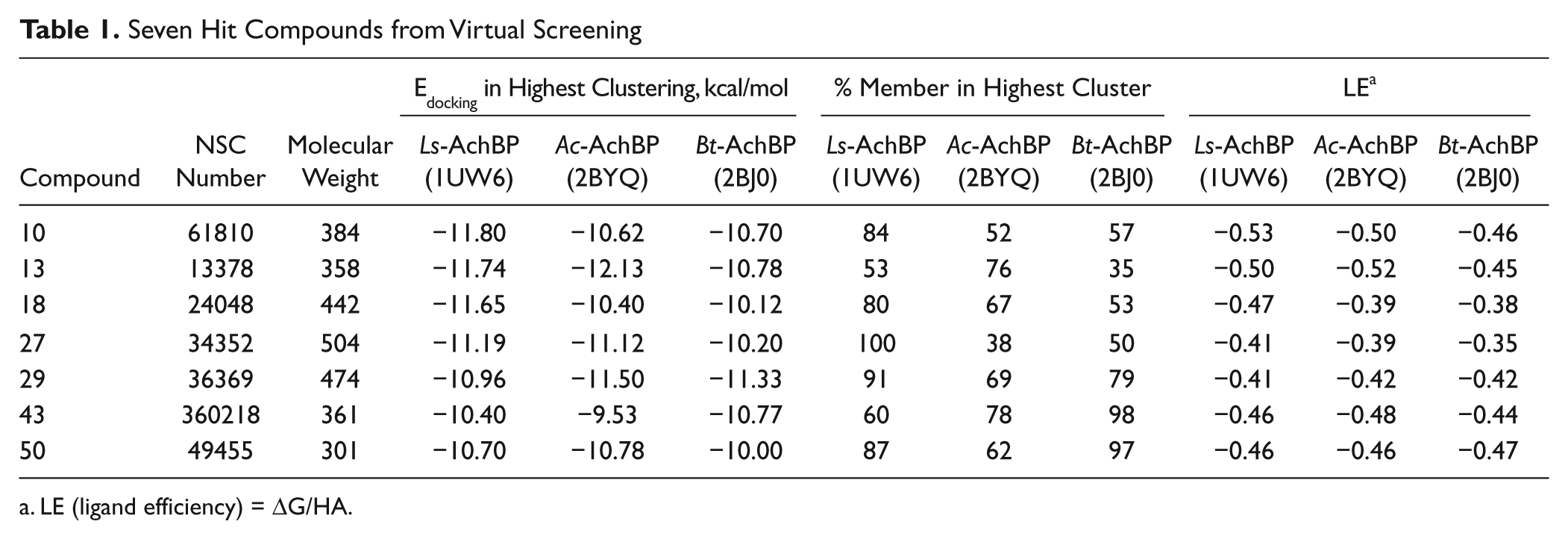
LE (ligand efficiency) = ΔG/HA.
Effect of hits on the nicotinic acetylcholine receptor binding
Fifty compounds from virtual screening were further investigated for their binding capability. The radioligand competition assay was adapted to determine whether the hits interfered with the binding of toxin to the nicotinic acetylcholine receptor. Prior to the binding analysis, 10 µM of each compound was screened for its ability to compete or interfere with the binding of [3H] epibatidine to the three AChBPs (Ls, Ac, and AcY55W), as measured by a scintillation proximity assay ( Fig. 1 ). Seven hits (compounds 10, 13, 27, 29, 40, 43, and 50) showed significant interference for the binding of [3H] epibatidine to BPs. These potential hits and their Kd values were measured by competing with [3H] epibatidine. A measured fraction greater than 1.0 is possibly due to a variation in the number of AChBP molecules binding to the beads.
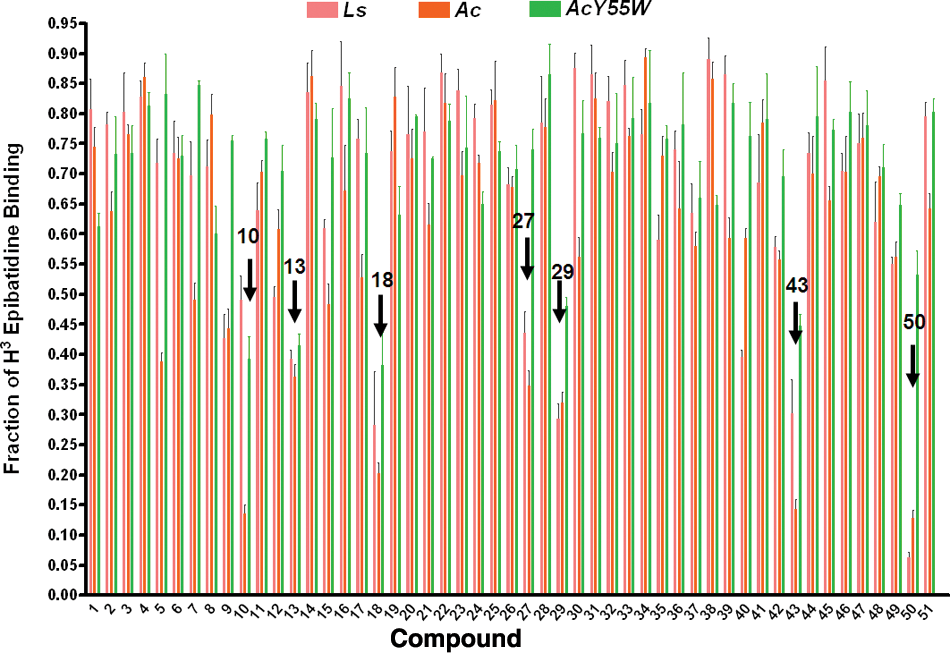
Screening test for the ability to displace the binding of [3H] epibatidine on Lymnaea stagnalis (Ls), Aplysia californica (Ac), and A. californica mutant (AcY55W). The x-axis is the log concentration of the compound at 10 µM; the concentration of [3H] epibatidine was 5 nM for Ls and AcY55W and 20 nM for Ac.
In the assay, the seven hits competitively displaced the agonist ([3H] epibatidine) from their mutually exclusive binding sites on Ls, Ac, and AcY55W AChBPs with the concentrations from micromolar to nanomolar (
Table 2
). The compound that bound more strongly to the BPs had a lower Kd value. NSC34352 (
Kd of Seven Hits for AChBPs
Average = dissociation constant value in nanomolar; n = number of the test.
All hit compounds except NSC34352 and NSC61810 are more drug like because they have a molecular weight less than 500, a log P less than 5, and fewer than five rotatable bonds (i.e., they each obey Lipinski’s rules) with a good LE 31 value between −0.35 and −0.53. LE is calculated by using binding free energy (ΔG) and number of heavy atoms (HA), LE = ΔG/HA. The docked poses of seven hits in three AChBPs are shown in Figure 2 , and the interacted amino acid residues are listed in Table 3 .
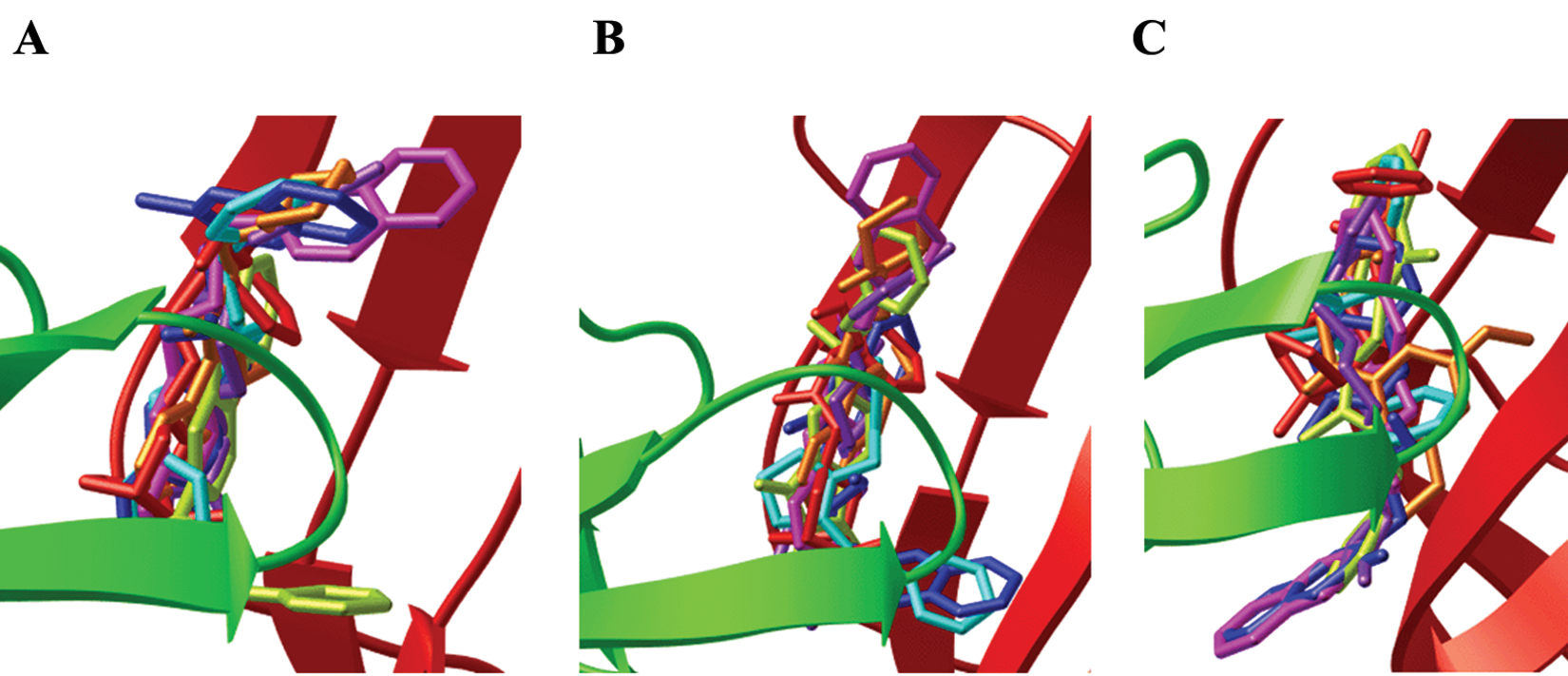
The docked orientations of AChBP interacted with the seven hit compounds from 1UW6 (
Intermolecular Interactions
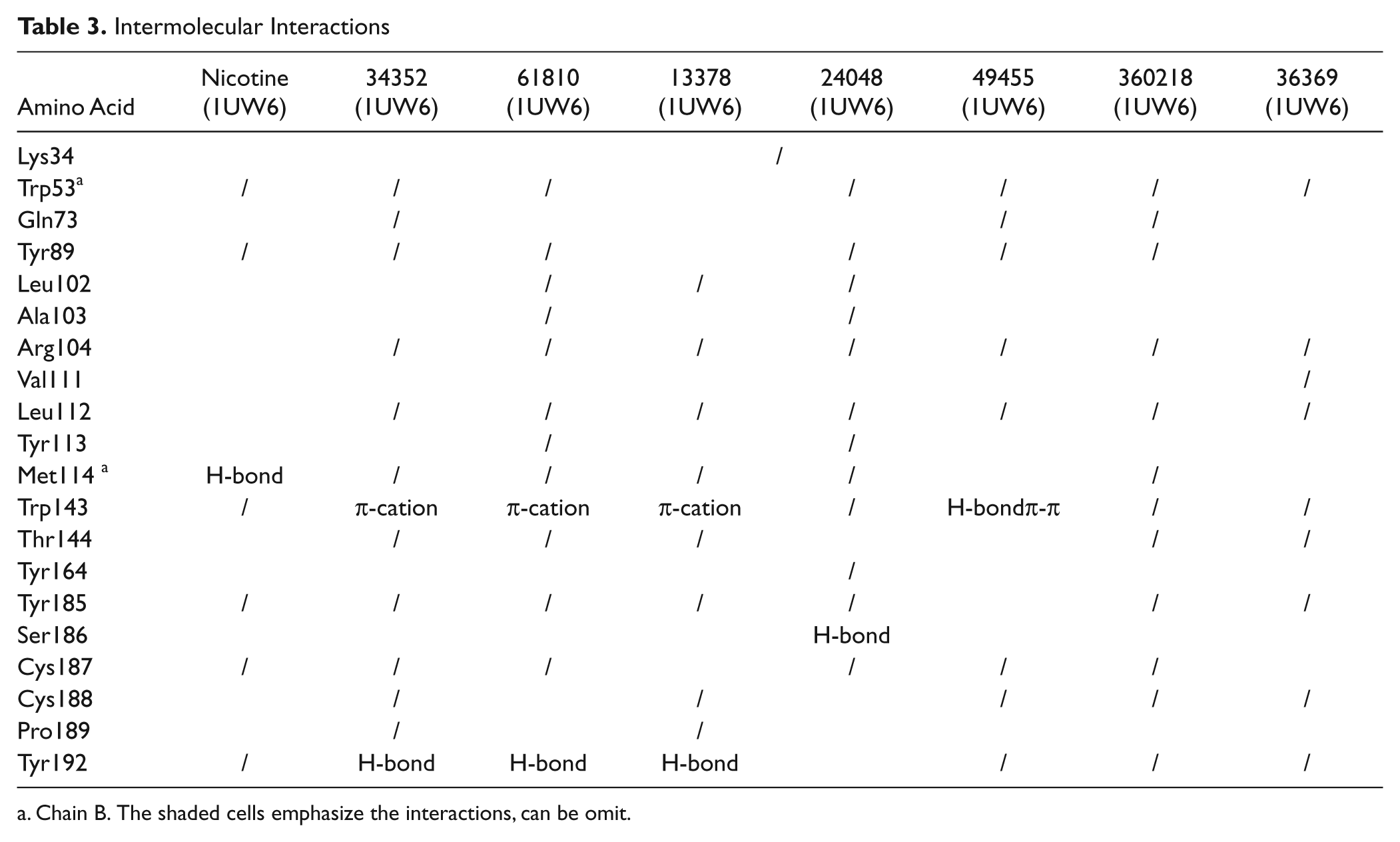
Chain B. The shaded cells emphasize the interactions, can be omit.
All hit compounds bound AChBP in the active binding site, with the docked poses located in the ligand-binding pocket at the interface of two adjacent AChBP subunits, which were the principal face in loop C and the complementary face in loop F. All ligands interacted with the major amino acid residues comprising an aromatic cage—that is, Trp53, Tyr89, Trp143, Tyr185, and Tyr192 ( Table 3 ). As expected, the hit compounds interacted with Trp143, Trp185, and either Cys187 or Cys188 of the disulfide bridged loop, which are the key pharmacophoric features of the α7nAChR agonist. The hit compounds that possessed aromatic functionalities were stabilized in the C-loop region by an aromatic cage; NSC61810, NSC13378, and NSC34352 also featured the chemical moieties bearing a charge and thus formed cation–π interactions with Trp143 in the binding site. The interactions between ligands and Trp143 varied from H-bonding, cation–π, and π–π. The structural components and their sizes played important roles in the orientation of the docked pose and the interacted distances, leading to variation in strength of binding and type of intermolecular interaction. The variations in receptor affinity and the occupied size resulted in conformational changes in loops C and F, especially in the region of the loop C tip, composed of residues Tyr185–Tyr192 and the disulfide bridge or the flexible flap or lid. The α7 agonist binding induced conformational change of loop C from open to closed and subsequently coupling of rotation in the inner region to the channel opening, whereas the antagonist binding contributed to the open conformation of loop C or the resting state. As the channel opening may entail only small concerted structural rearrangements that are difficult to predict by docking and the result from the radioligand competition assay could not differentiate between agonist binding and antagonist binding, the functional assays using α7nAChR expressed in Xenopus oocytes were conducted to determine whether the hit compounds are agonists or antagonists. Among seven identified hit compounds, the structures of NSC61810, NSC34352, and NSC36369 are very similar to those of anticholinergic drugs. The tropane-bearing structure of NSC61810 is similar to antimuscarinic ipratopium, whereas NSC34352 and NSC36369 contain a bisonium function the same as antinicotinic pentolinium and hexamethonium. However, we expected that the minute differences in structure or orientation would alter the mode of action from antagonist to agonist.
The functional assay using α7nAChR expressed in Xenopus oocytes
First, the pharmacological property of α7nAChR expressed in Xenopus oocytes was characterized. When ACh 10 to 1000 µM was bath applied to oocytes injected with α7nAChR cRNAs, inward currents were elicited at holding potentials of −80 mV. The current response elicited by 100 µM ACh was completely inhibited by the competitive antagonist MLA 1 µM (data not shown). Concentration–response curves for ACh were made by applying different concentrations of ACh. The 50% effective concentration (ED50) values and correlation coefficients for curve fitting were determined using the Prism (version 2.0) program. Maximal currents were observed at 1000 µM, and the EC50 value was 179 µM. In further experiments, we used 100 µM ACh as a standard concentration.
To investigate whether the compounds were agonists or antagonists, the compounds were bath applied to the oocytes expressing α7nAChR for 1 min. None of the compounds elicited an agonistic effect to α7nAChR up to 300 µM. We have further determined the antagonist effect of the hit compounds at a concentration of 100 µM on the current induced by ACh (100 µM). All hit compounds inhibited ACh-induced currents by 69% to 100% (n = 4, SD < ±5%) of control, respectively. The antagonist effect was found to be dose dependent, and the inhibitory effect of the compounds disappeared after washing out of compounds by buffer for 5 min, indicating that the compounds are the reversible antagonists of α7nAChR. The result of this study showed that all hit compounds inhibited the ACh-mediated response of α7nAChR expressed in Xenopus oocytes in a manner characteristic of reversible antagonists. Despite a high inhibitory effect on ACh-induced current, the effect of these compounds was less potent than that of MLA.
To investigate whether the compounds were agonists or antagonists, the compounds were bath applied to the oocytes expressing α7nAChR for 1 min. None of the compounds elicited an agonistic effect to α7nAChR up to 300 µM. We have further determined the antagonist effect of the compounds at a concentration of 100 µM on the current induced by ACh (100 µM). All hit compounds inhibited ACh-induced currents by 69% to 100% (n = 4, SD < ±5%) of control, respectively ( Fig. 3 ). The inhibitory effect of the compounds disappeared after washing out of compounds by buffer for 5 min, indicating that the compounds may be the reversible antagonist of α7nAChR. The result of this study showed that all hit compounds inhibited the ACh-mediated response of α7nAChR expressed in Xenopus oocytes in a manner characteristic of reversible antagonists. NSC49455 showed the highest inhibitory effect on ACh-induced current, but the effect of these compounds was less potent than that of MLA.
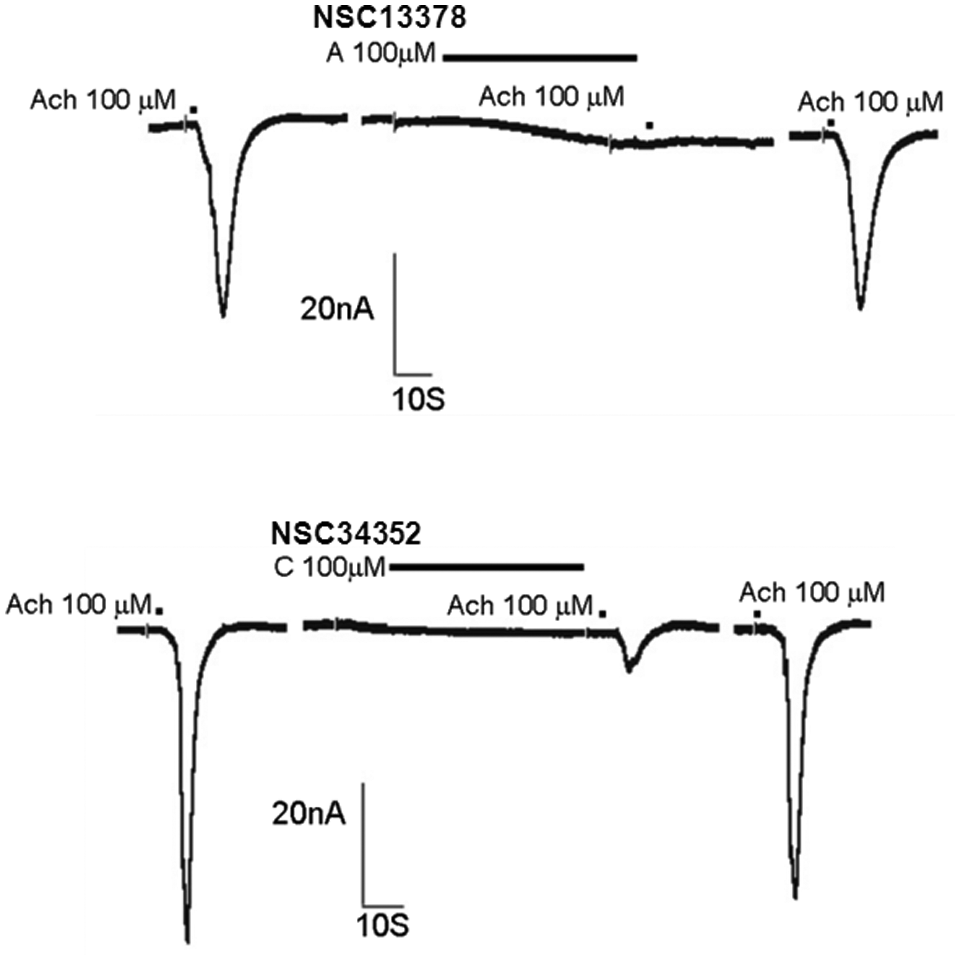
Inhibitory effect of hit compounds on acetylcholine (ACh)–induced current in Xenopus oocytes expressing α7nAChR. Current recorded from a representative oocyte that expressed α7nAChR, illustrating the effect of the hit compounds (100 µM) on currents evoked by 100 µM ACh. The oocyte was voltage clamped at −80 mV. ACh was applied for 7 s with or without the compound pretreatment for 2 min. The drug was then washed out for 5 min, and ACh was applied again.
The functional assay using recombinant receptors and FRET-based calcium sensor expressing in HEK cells
Seven hit compounds from the functional assay were investigated further for selectivity by the functional LGIC–CNiFER assay.
36
The assays were performed on α7nAChR CNiFERs, α4β2nAChR–CNiFERs, and 5-HT3A CNiFERs. To assess agonist activity, each compound was first added to the well at a final concentration of 10.3 µM. Interestingly, NSC34352 (
Functional Activities on LGIC–CNiFERs a
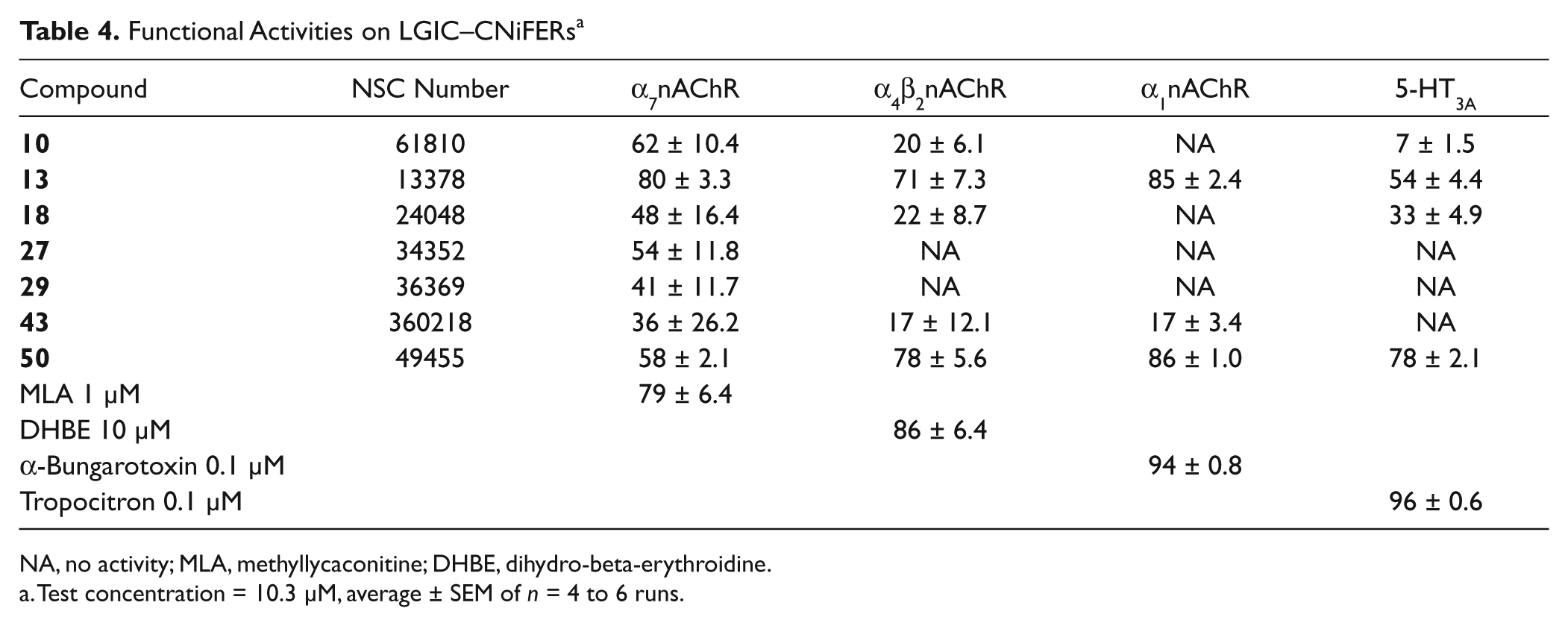
NA, no activity; MLA, methyllycaconitine; DHBE, dihydro-beta-erythroidine.
Test concentration = 10.3 µM, average ± SEM of n = 4 to 6 runs.
According to the functional assay, all hit compounds had an effect opposite that which was expected: antagonistic instead of agonistic. As indicated from the observed data shown in Table 3 , the antagonistic effect may have arisen from the H-bond interaction with Tyr192 or the bulky and rotatable side chain, forcing the functional loop C open ( Fig. 4 ). The most potent antagonistic effect of NSC13378 resulted from the rotatable (piperidin-2-yl)ethyl side chain, leading to the open configuration of the loop C flap. The weakest antagonist was NSC360218 due to its lack of an H-bond formation. NSC34352 was the only hit compound that exerted not only an antagonistic effect but also a partial agonistic effect. The antagonistic effect can be attributed to the steric methylisoquinolinium it is not quinoline as the quinolines in the structure of NSC34252 are quaternary ammonium ions. moiety and the H-bond interaction, causing loop C to open or partially open. The methylisoquinolinium was aligned parallel to the indole of Trp143, forming a cation–π interaction with optimal force to couple binding to gating ( Fig. 3 ).
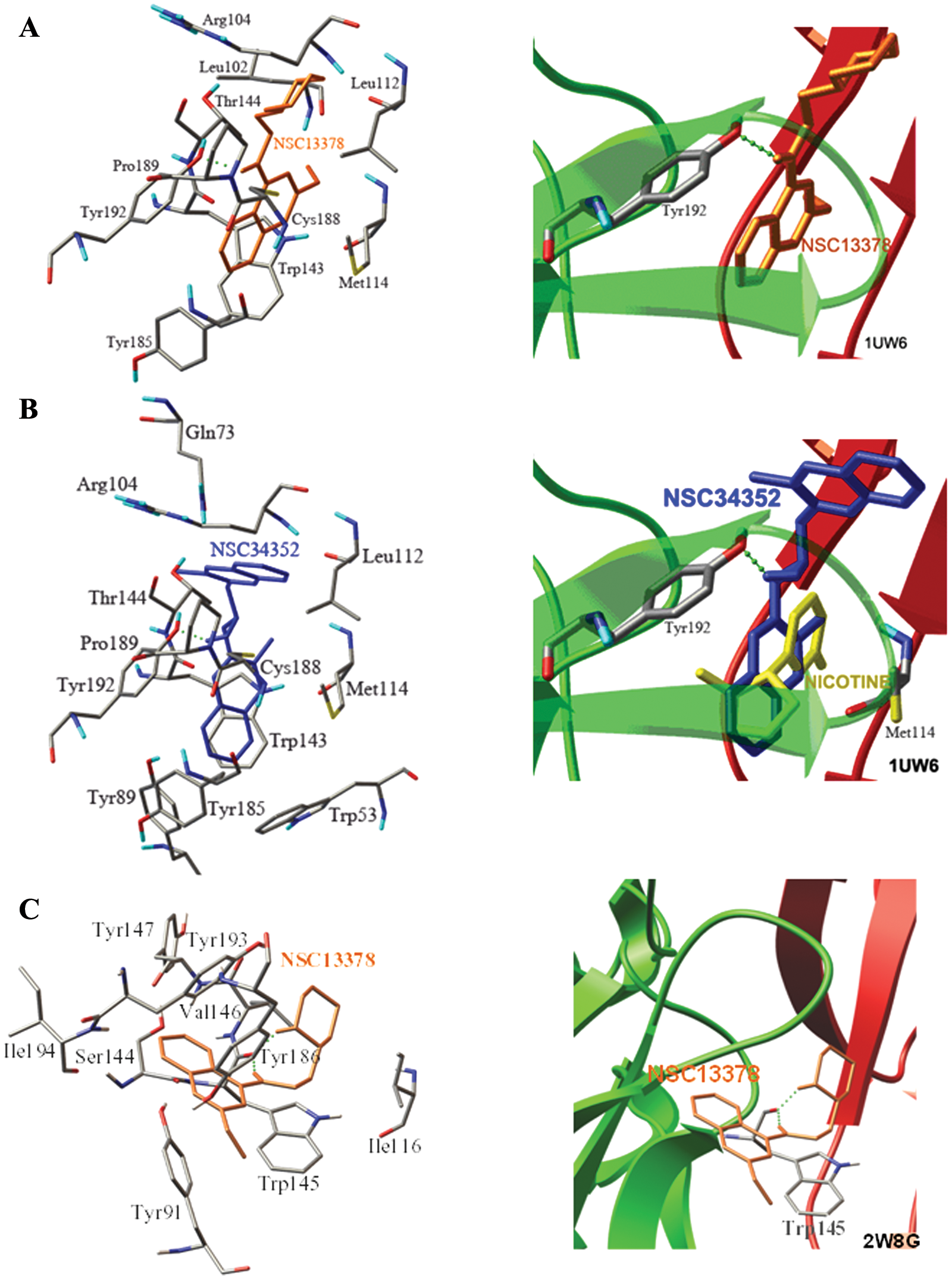
The binding modes of the α7 antagonists, NSC13378 (
Dockings of these hit compounds to the AChBP model derived from the antagonist-bound Ac-AChBP complexes (2W8G 38 ) were tried to differentiate between agonist and antagonist bindings. The docking result demonstrated that all hit compounds were located in the aromatic cage beside loop C, and the binding affinities were comparable to those from the agonist-bound Ls-AChBP (1UW6). Although the docked orientations of the ligands and the amino acid residues forming the H-bond were different, the differences found in seven compounds did not have the same trend. The obtained results are not sufficient, and it is complex to distinguish between agonists and antagonists. For instance, the binding poses of NSC13378 when docking to the AChBP templates from 1UW6 ( Fig. 4A ) and 2W8G ( Fig. 4C ) were considered different (RMSD > 2Å), and the amino acid residues forming the H-bond were Tyr192 for 1UW6 and Val146 for 2W8G. The binding affinities of NSC13378 were of comparable magnitude (–11.74 kcal/mol and −10.29 kcal/mol).
Although the hit compounds did not exert the potent agonistic effect as anticipated, a number of findings have demonstrated the potential effects from antagonizing α7nAChR in the treatment of Alzheimer disease. It has been recently shown that Aβ binds to α7nAChR and mediates Aβ-induced tau protein phosphorylation, and inactivation of functional α7nAChR improves memory and cognitive deficits in the APP-overexpressing mouse.40–42 Thus, these findings suggest that blocking the α7nAChR function with an antagonist could be beneficial in the treatment of Alzheimer disease. The information from the screening, regardless of the very small diversity library (1990 compounds), is satisfactory, and the core structures of
Virtual screening is increasingly gaining acceptance in the pharmaceutical industry as a cost-effective and timely strategy for analyzing very large chemical data sets for potential interactions with therapeutic targets. Although the number of therapeutic targets that have been fully characterized by crystallography is currently limited, this situation is changing significantly as structural genomics initiatives begin to yield fruit. Accordingly, the work involved to validate all these potential targets, demonstrate their therapeutic relevance, and find effective ligands will become more dependent on the new high-throughput screening technologies. Molecular docking was used to investigate the binding of more than 1990 compounds to AChBP. This procedure is computationally intensive for analyzing a large database but provides the most detailed basis for determining which compounds are likely to be potential ligands. Seven hits showed good competitive activity in interfering with the binding of [3H] epibatidine to the AChBPs and elicited antagonistic activity on α7nAChR. NSC36369 and NSC49455 showed good competitive activity, but only NSC34352 elicited partial agonistic activity on α7nAChR. Nevertheless, these novel hits could be helpful in pharmaceutical research on various neurological processes and anticancer therapy.
Footnotes
Acknowledgements
This work was supported by the Commission on Higher Education (grant number CHE-RG-2551-53, NRU-PY540105), Ministry of Education, Thailand, and the JSPS-NRCT Core University Program 2009. Computer modeling resources were provided by the Molecular Graphics Laboratory at The Scripps Research Institute. The authors thank John G. Yamauchi and Ákos Nemecz for assistance and suggestions with regard to the functional LGIC–CNiFERs assay.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.