
Abstract
Bromodomains are structurally conserved protein modules present in a large number of chromatin-associated proteins and in many nuclear histone acetyltransferases. The bromodomain functions as an acetyl-lysine binding domain and has been shown to be pivotal in regulating protein–protein interactions in chromatin-mediated cellular gene transcription, cell proliferation, and viral transcriptional activation. Structural analyses of these modules in complex with acetyl-lysine peptide ligands provide insights into the molecular basis for recognition and ligand selectivity within this epigenetic reader family. However, there are significant challenges in configuring assays to identify inhibitors of these proteins. This review focuses on the progress made in developing methods to identify peptidic and small-molecule ligands using biophysical label-free and biochemical approaches. The advantage of each technique and the results reported are summarized, highlighting the potential applicably to other reader domains and the caveats in translation from simple in vitro systems to a biological context.
Keywords
Introduction
D
What are bromodomains?
Bromodomains (BDs) are small (~110 AA) evolutionary conserved modules present in a large number of chromatin-associated proteins and in nearly all nuclear histone acetyltransferases. They were first identified as a motif in Drosophila Brahma from where their name arose 9 but were also soon after located in humans and yeast. Most commonly found as single copies, frequently in combination with other epigenetic reader domains such as plant homeodomain (PHD) and proline-tryptophan-tryptophan-proline (PWWP), these concatenated domains are believed to act in a synergistic manner to confer specificity for the complex pattern of modifications known as the “histone code.” In cases where multiple bromodomains occur within a protein, they are contiguous (e.g., bromodomain and extra-terminal domain [BET] family of proteins, 10 human polybromodomain-1 [PB1]11,12), and domains within the same protein often exhibit no greater similarity to each other than to BDs in other proteins, suggesting that the individual domains may possess a distinct function. The human genome encodes for 56 distinct bromodomains contained within 42 proteins.
Emerging therapeutic potential of bromodomains
Human BD-containing proteins have been implicated in a wide range of diseases, including oncology, inflammation, obesity, diabetes, infectious diseases, and metabolic and cardiovascular indications.13,14 Most of these disease linkages have been generated from correlating expression patterns, phenotypes following gene knockout or knockdown, and genetic associations. However, deconvoluting the specific relevance of the bromodomain within these proteins to disease processes has been difficult without access to tool compounds, and therefore the therapeutic potential of this target class remains to be fully elucidated. In the context of this review, we have chosen to concentrate on proteins where the emerging data from small-molecule inhibitors and site-directed mutagenesis experiments clearly demonstrate the importance of the bromodomain itself rather than that of the entire protein. Despite this rather selective focus, the potential of bromodomain inhibitors to affect many areas of biology is readily apparent.
The BET bromodomain family of proteins plays an essential role in governing cell fate and cell cycle progression. 10 Imbalance between cellular decisions to grow, specialize, or undergo apoptosis is central to oncogenesis,13–15 and the link between BET proteins and some human cancers is now compelling. A clear example stems from the work of French and colleagues.16,17 They studied the incidence and pathology of a rare, but invariably lethal, midline carcinoma caused by the translocation of the BRD4 gene and, more infrequently, the BRD3 gene, with the nuclear protein in testis, NUT. This results in the abnormal expression of a fusion protein consisting of the tandem N-terminal bromodomains of BRD3/4 as an in-frame chimera with NUT. Oncogenesis is believed to arise as a consequence of this aberrant protein, sequestering p300 HAT to BRD-NUT-acetyl-chromatin sites, causing overabundant HDAC activity outside these regions and thus global histone hypoacetylation and transcriptional repression of a range of tumor suppressor genes.18,19 In cell lines carrying the translocation, siRNA to the BRD4 protein was sufficient to both reduce proliferation and promote cellular differentiation into keratinocytes. These in vitro effects were echoed by the application of the small-molecule pan-BET inhibitor, JQ1, which was also able to significantly inhibit tumor growth in two mouse xenograft models of the NUT midline carcinoma. 5
Another area where the importance of the BET bromodomains has been demonstrated is in regulating inflammatory responses. Endotoxin-mediated activation of the innate immune system, using lipopolysaccharide (LPS) stimulation of Toll-like receptor 4 (TLR4), has been shown to involve BRD4 regulation of the NFkB-dependent genes. 20 BRD4 has also been found to regulate inducible gene expression in macrophage primary response genes. 21 A detailed study of the small-molecule Bet inhibitor, I-BET,1,22 highlights its ability to suppress a subset of LPS-induced genes, the secondary response genes, in mouse bone marrow–derived macrophages by selectively preventing the recruitment BRD2, 3, and 4 to these specific gene promoters. The translation of these in vitro observations into an in vivo setting of a mouse model of endotoxic shock was striking. Prophylactic and therapeutic dosing of I-BET suppressed cytokine expression and promoted survival in mice administered with lethal doses of LPS. Compounds from this same inhibitor class have also been reported as upregulators of ApoA1, which supports their potential use as anti-inflammatory, metabolic, and cardiovascular agents. 22 Although a body of literature suggests a role of BET proteins in the life cycle of infectious agents, including human immunodeficiency virus (HIV), herpes, and papilloma viruses,23,24 both the bromodomains and the ET (extra-terminal) regions have been implicated in transcriptional regulation. 25 Therefore, the therapeutic utility of targeting the bromodomain alone for infectious diseases remain to be established.
A number of studies suggest that the interaction of the bromodomain of PCAF (p300/CREBBP-associated factor) with the HIV Tat protein is central to efficient HIV replication.26,27 This interaction is dependent on the acetylation of K50 of Tat. 26 Small-molecule inhibitors that block the acetyl-lysine binding to PCAF present an opportunity to halt HIV progression that mitigates some of the problems due to drug resistance. 4
The tandem ATPase and bromodomain-containing protein ANCCA (AAA nuclear coregulator cancer-associated protein) or ATAD2 was identified as a direct target of the oncogene AIB1/ACTR/SRC-3 and is strongly implicated in the tumorigenesis of several cancers.28,29 ANCCA is overexpressed in >70% of breast tumors, and protein levels are correlated with disease aggression, tumor metastasis, and poor survival rates. 30 ANCCA is known to control the expression of B-Myb, EZH2, and Rb-E2F, along with a subset of key mitotic kinesins and cell survival genes. Recent mutagenesis studies of the bromodomain of ANCCA indicate it serves a critical role as a chromatin anchor, assisting the effective recruitment and assembly of E2Fs and other complexes that mediate cell cycle genes. 31 Inhibiting bromodomain binding would therefore diminish the ability of ANCCA to act as an E2F coactivator and promote cancer cell proliferation.
The human transcriptional coactivator CREBBP (CREB binding protein) has an extensive spectrum of protein partners and bridges many DNA binding transcription factors to the basal transcriptional machinery. 32 One such partner is the tumor suppressor p53. Upon DNA damage, p53 becomes acetylated on K382, which associates with the bromodomain of CREBBP, resulting in transcription activation of p53 target genes. 33 Recent studies indicate that CREBBP is a proto-oncogene involved in tumor initiation, progression, and metastasis. 34 Elevated levels of CREBBP are observed in patients with prostate, breast, and non–small cell lung cancers, as well as acute leukemia. In contrast, downregulation of CREBBP in several cancer cell lines results in reduced cell proliferation and induction of apoptosis. Together, this suggests CREBBP may be an alternative to direct p53 inhibition as a target for cancer therapy. Excess p53 activity has also been linked to neurodegenerative disorders,35,36 infectious 35 and autoimmune disease, and ischemic heart disease. 37 CREBBP inhibitors may therefore have benefit in these areas. An initial indication of this potential is demonstrated by the discovery of small-molecule inhibitors of the CREBBP bromodomain that are able to modulate the effect of p53 in vitro,2,38 but effective translation into cellular and in vivo settings ideally requires more optimized molecules.
Structure of bromodomains
The first structure of a bromodomain, that of PCAF, was revealed by nuclear magnetic resonance (NMR) in 1999. 39 Over the following decade, structures of more than 20 additional members of this family have been solved by X-ray and NMR. These all share a common fold, consisting of four antiparallel α-helices (αZ–αA–αB–αC) arranged in a left-handed twist ( Fig. 1 ). Forty-four of the 56 bromodomains possess a conserved tyrosine and asparagine that form the acetyl-lysine binding motif as highlighted in the sequence alignment shown in Figure 2 . The remaining bromodomains have an atypical recognition signature (e.g., PB1–BD4/6) whose functional consequences are unknown. The insert of Figure 1 illustrates the structural basis for acetyl-histone peptide binding within a “typical” bromodomain. The carbonyl of the acetyl group makes both a direct hydrogen bond with the conserved asparagine and a water-bridged interaction with the conserved tyrosine, whereas the methyl group of the acetyl sits in a small hydrophobic pocket. In some bromodomains (e.g., BRD4), a larger propyl moiety can be accomodated. 40 The biological relevance of this tolerance is unclear, but histones containing both propionylated and butyrylated lysine residues have been found in yeast and mammals. The charge of an unmodified lysine would sit uncomfortably within this largely hydrophobic environment, explaining the preference for a neutralizing modification. The long ZA and shorter BC loops that flank the Kac recognition site are the most variable regions between the bromodomains, consistent with their role in discriminating Kac in the context of different peptide sequences. Although the main function of these proteins is believed to be histone related, bromodomains have also been implicated in binding to other acetylated proteins such as RelA, 20 p53, 41 and HIV Tat. 26
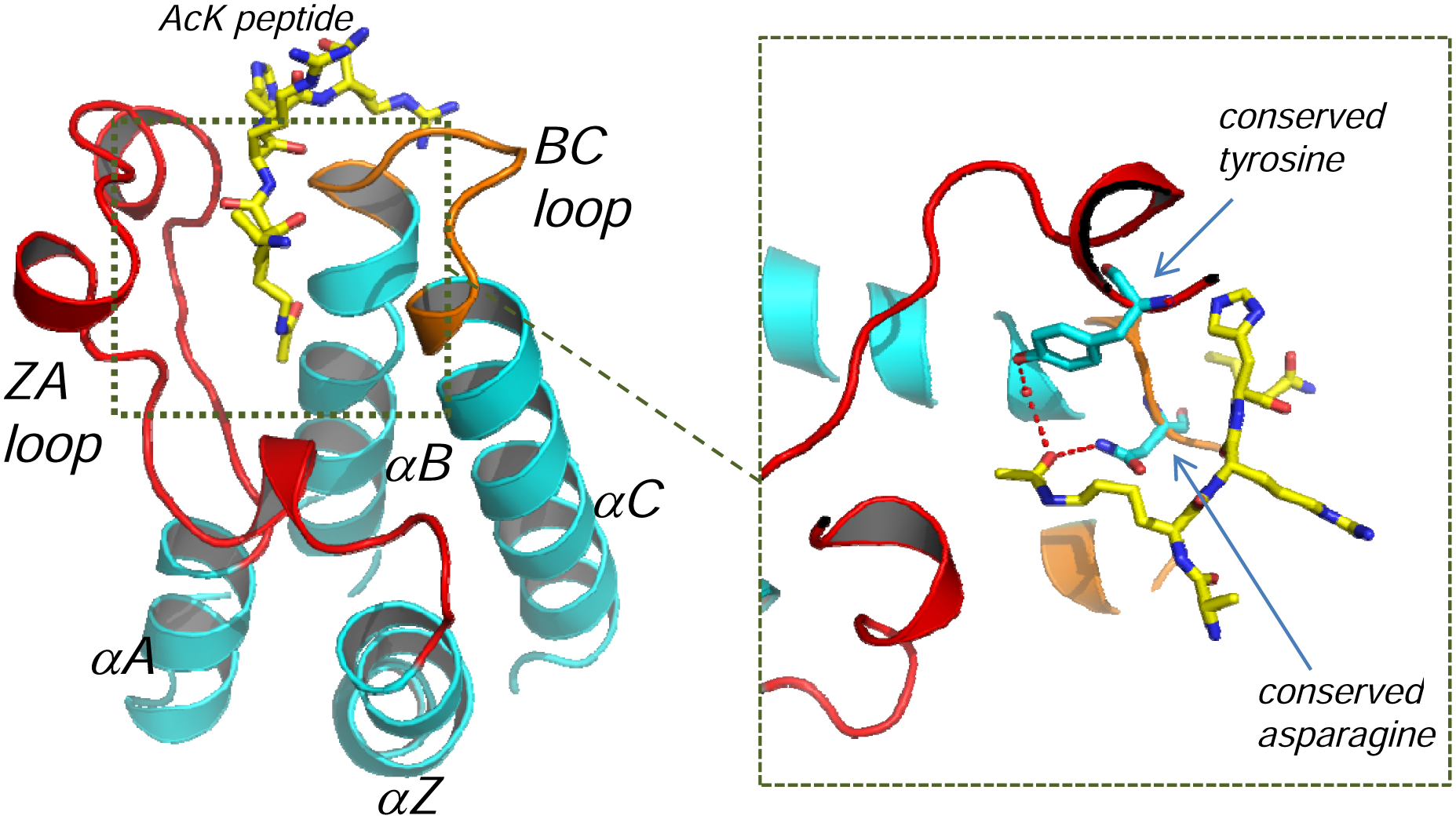
Bromodomain structure and Kac recognition. The archetypal four-helix bundle topology of bromodomains (αZ–αA–αB–αC) is illustrated by the structure of BRD2–BD1 (cyan) complexed to the H4K12ac peptide (yellow) (PDB entry 2DVQ). Highlighted in red and orange are the ZA and BC loops that flank the acetyl-lysine binding pocket. Variability in these loops allows the Kac residues to be recognized in the context of differing peptide sequences. The insert shows the conserved tyrosine and aparagine in stick format and the hydrogen bonding network within the Kac binding site to the carbonyl of the acetyl.

Sequence alignment of 12 bromodomains. The secondary structural elements (gray) and conserved recognition tyrosine (black) and asp (dark grey) are highlighted. The atypical bromodomains of the sixth and first bromodomains of human polybromodomain–1 are shown (PB1–BD4/6). The sixth lacks the conserved tyrosine, and the first has a tyrosine in place of the asparagine.
Most structures determined are of the apo protein, as relatively few peptide and even fewer small-molecule ligands of low micromolar affinity are known. Consequently, although the overall architecture and critical requirements for substrate recognition are well established, our detailed understanding of the elements necessary to achieve high-affinity interactions is only beginning to emerge.
Challenges for screening bromodomains
Bromodomains possess no enzyme activity; their function is mediated solely via protein–protein interactions. For the majority of bromodomains, there is little definitive knowledge about their cognate sequences, and few tool compounds have been identified. Configuring and validating a suitable assay to identify Kac inhibitors without competitive tool molecules to confirm the performance of a screening format is demanding and requires careful consideration of both the primary screen and the subsequent triaging cascade.
The recent revelation of the tractability of this target class to small-molecule inhibition means that there are limited screening data published to date. In vitro screening options attempted thus far can be conveniently divided into two distinct approaches: those that monitor binding directly using a label-free (LF) methodology (e.g., NMR, surface plasmon resonance, differential scanning fluorimetry, etc.) and those that detect the displacement of a partner protein, peptide, or small molecule (e.g., enzyme-linked immunosorbent assay [ELISA]–based formats, fluorescence anisotropy, time-resolved fluorescence resonance energy transfer [TR-FRET], AlphaScreen, etc.).
In this review, we give examples where these technologies have been used to screen for bromodomain inhibitors. We also describe instances where techniques have been applied in a bespoke fashion to characterize small numbers of molecules but where there is potential for more expansive hit identification usage. In addition, technologies that have demonstrated utility for other reader domains that can be similarly employed for bromodomains are cited. Finally, extensive biophysical studies have been used to validate binding partners. Some of these methods, such as isothermal titration calorimetry (ITC) and X-ray crystallography, will also be mentioned as, for some targets, these may be one of the definitive ways of unraveling the mode of action of peptide and compounds. This is especially the case for the atypical bromodomains and for bromodomains that can be screened only as part of larger multidomain constructs where multivalent interactions with the binding partner may complicate interpretation.
Label-Free Direct Binding Assays
The major advantage of using an LF format is that it negates the need to identify a suitable peptide or protein partner. However, normally all binding events are detected, and therefore triaging to determine specificity and functional relevance is key. High-content techniques such as protein NMR and surface plasmon resonance (SPR) are attractive as they capture information that can be used to eliminate compounds with an undesirable mode of action (MOA) at an early stage. Neither of these formats is amenable to true high-throughput screening (HTS). Therefore, it is fortunate that the structural characterization of at least the typical bromodomains suggests that a target class approach, using comparatively small focused compound sets, may be sufficient to generate suitable starting points in the absence of a full diversity screen. Other biophysical methods such as differential scanning fluorimetry (DSF), ITC, and X-ray crystallography have also been used as part of the hit confirmation strategy, and the additional value they offer will be stated. It should be noted that label-free plate-based platforms 42 could, in principle, be used for higher throughput screening for epigenetic reader proteins. No examples are known to date, but reference to the considerations for other LF approaches should inform the feasibility of their usage.
Nuclear magnetic resonance
Thus far, the LF approach that has been most successfully applied to identify and characterize small-molecule bromodomain inhibitors is protein NMR, using 2D 15 N-heteronuclear correlation spectroscopy (e.g., 1H–15N HSQC). This has largely been the work of the group of Ming-Ming Zhou,2–4,38 who has focused on the bromodomains of two HAT proteins, PCAF and CREBBP. Both these proteins are known to associate with acetylated histones, but they have also been shown to interact with acetylated HIV Tat (K50ac) and p53 (K382ac), respectively. Antagonizing these nonhistone interactions would interfere with HIV viral replication in the case of PCAF and with tumor growth for CREBBP.
1H-15N NMR screening has been widely used within the fragment arena and was the archetypical experiment reported by the Abbott group in their seminal SAR-by-NMR work on this subject.43,44 The method involves running heteronuclear 1H–15N correlation experiments in the presence and absence of a ligand or pool of ligands and monitoring the differences within the spectra, as exemplified schematically in Figure 3a . Each resonance in a 1H–15N HSQC spectrum represents a distinct H–N pair, which means that the spectrum gives a residue-resolved map of the protein backbone and has high and easily interpretable information content. The specific binding of a ligand results in a subset of peaks in the spectrum moving (chemical shift change) and/or disappearing (line broadening). This allows not only the presence of an interaction to be detected but potentially also the site of interaction to be mapped. This can be achieved by knowledge of the full-residue assignment of the individual peaks or by use of a known binder, which can be very weak (e.g., Kac), to locate the subset of peaks corresponding to the site of interest. Undesirable modes of action such as protein denaturation or nonspecific effects can also be gauged by the appearance of the spectra after ligand addition.
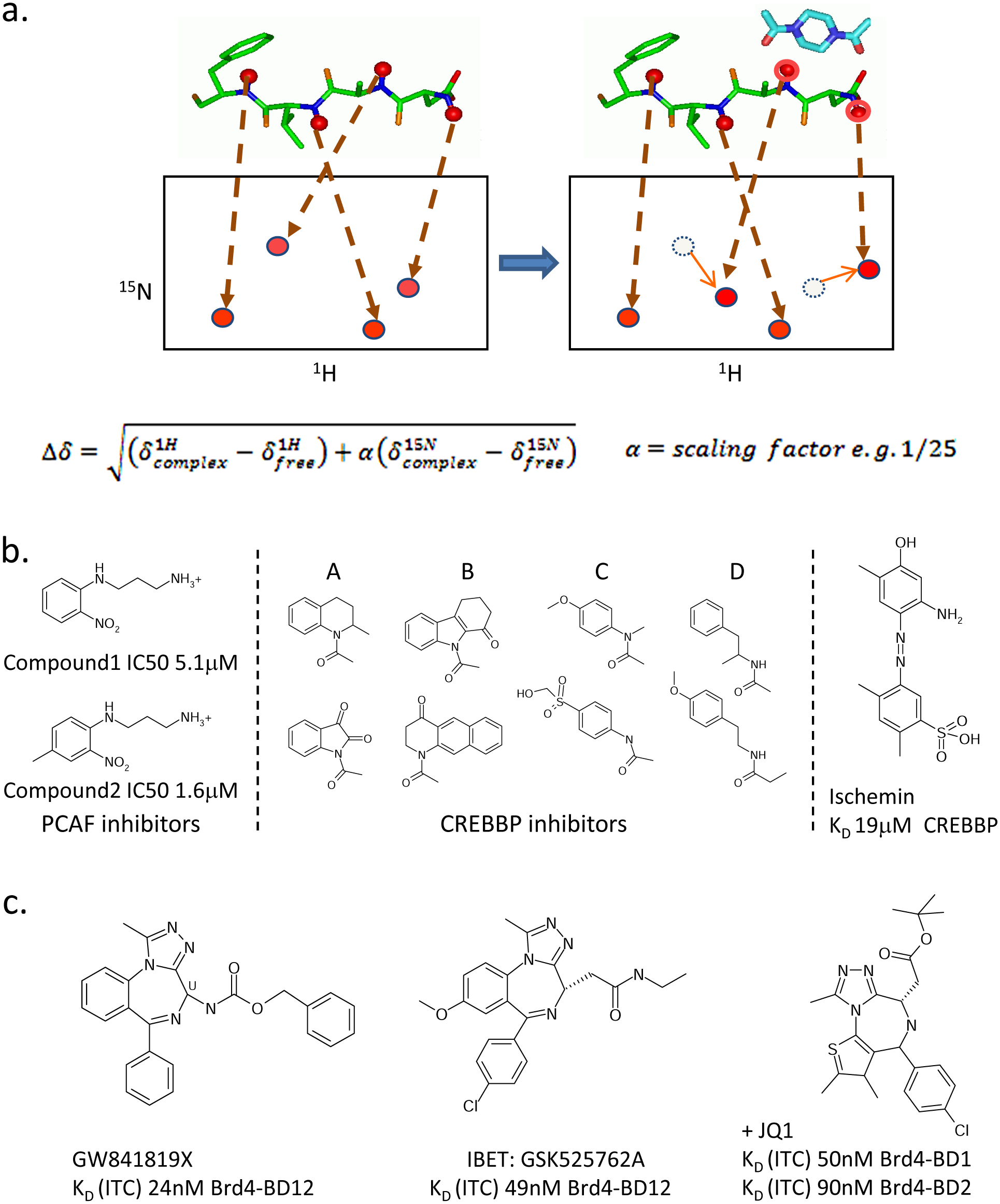
Nuclear magnetic resonance (NMR) screening and compounds discovered. (
Although this method certainly cannot be considered high-throughput, it has many attributes that make it both an attractive and viable option for screening thousands of compounds against bromodomains. First, isolated bromodomains are typically well expressed in Escherichia coli (~10 mg/L), and the generation of 15 N material is usually feasible and cost-effective using minimal media containing 15 NH4 chloride or sulfate salts as the sole nitrogen source. Second, the initial appearance of the spectrum can confirm the integrity of the proteins—an important consideration when small truncates are taken out of context of considerably larger proteins and a physiological environment where they may be stabilized in multimeric complexes, but where in isolation they possess no easily measurable enzymatic activity. Third, this method’s large dynamic range (from millimolar to nanomolar), coupled with the ability to confirm a desirable MOA and the binding site, gives highly validated hits that are invaluable for targets where there are few other tools available for confirmation. Last, although resource intensive, this technique can be used to determine both the affinity of binding and a full 3D structure if required. Affinities are typically measured by monitoring the gradual chemical shift perturbations (as defined in Fig. 3a ) as a function of sequential titration of the ligand into a sample and fitting this change to a standard binding equation. Indeed, for very weak ligands (e.g., >100 µM), the HSQC’s ability to monitor site-specific events may make it the only viable way of differentiating between concentration-dependent-specific and nonspecific binding.
The detectability limit of this NMR assay is dependent on both the precise chemical nature of the ligand and its binding mode, as only ligand-induced changes in the chemical environment of amide and side chain HN groups that are not exchange broadening are measured. The water-filled and side chain–rich Kac recognition site of bromodomains may render the cavity itself less sensitive than binding at the peripheries of this site, where more proximal and direct protein backbone amide interactions are possible.
PCAF 4 was the first bromodomain to undergo NMR screening. Compounds were screened using a variety of methods, including 1D NOE-pumping, 45 saturation transfer, and 2D 15 N–HSQCs. Focusing on selectivity, computational approaches were used to select the screening set of a few thousand commercial compounds designed to bind PCAF near, but not at, the Kac binding pocket, to exploit the differential nature of the ZA and BC loops between BDs. Several active molecules were found, including compound 1 ( Fig. 3b ), which had comparable affinity to the Tat peptide. The relative affinities of this compound and analogs were determined by ELISA using an immobilized biotinylated Tat K50ac peptide and glutathione (GST)–PCAF. The most active compound (compound 2), a methyl substitutent of compound 1, had an IC50 of 1.6 µM, threefold lower than compound 1. Members of this class of N-aryl-1,3-propane diamine compounds were shown to have no activity against BDs of CREBBP and TIF1β, demonstrating that relatively small molecules may show selectivity.
Following the success of this approach, the group turned their attention to CREBBP. 3 This time, a focused set of 200 compounds predicted to bind within the Kac site itself was chosen. The set of 200 was divided into 25 pools each with eight compounds and screened by 15 N-HSQC. Deconvolution of active pools yielded 14 Kac site actives, giving a hit rate of 7%. This high confirmed hit rate demonstrates the amenability of bromodomains to a structure- and fragment-based approach. Despite the fact that the set was focused on targeting the conserved Kac site, the hits exhibited a preference for PCAF-BD over CREBBP, reiterating the potential for differentiating interactions close to the recognition pocket. Most recently, this group has also published the discovery of azobenzene compounds ( Fig. 3b ) in the tens to hundreds of micromolar range for CREBBP also found by 15 N NMR screening and cyclic peptide mimetics of p53. 2
These examples illustrate both the power of an NMR approach and its limitations. For structure-based focused screening and fragment efforts using a few thousands compounds, this is undoubtedly an enabling and valuable method, which can yield validated starting points for chemistry. For routine SAR generation and where larger screening campaigns are required, the transformation of these initial tools to conventional competition formats is clearly the logical step, and examples of this will be given.
Surface plasmon resonance
SPR has become a popular technique for fragment studies and has the advantages of using far less protein, being higher throughput, and more easily generating affinity and kinetic data than NMR screening.46–49 This method immobilizes one component of the interaction onto the surface of a gold chip, over which solutions of putative binders are sequentially introduced. Although array-based SPR technologies have been reported,50,51 the most common implementation involves a continuous-flow microfluid system over which discrete plugs of analyte solution are introduced. The resulting sensorgrams monitor the interaction as a function of time and so capture the kinetics of binding as well as the degree of interaction with the surface. As the size of the response signal is proportional to the molecular weight of the analyte, the most sensitive and easiest assay configuration would place a biotinylated histone peptide onto the gold surface and test compounds by their ability to block the binding of a bromodomain that is co-injected over the surface. Unfortunately, this does not fully exploit the advantages of an LF approach as this is essentially a competition assay, and the only kinetics monitored will be those of peptide-histone binding. If, however, the protein can be immobilized onto the gold surface, then the binding of compounds can be directly observed. The resulting binding sensorgrams are much more information rich and yield telltale signs of nondesirable modes of action, such as abnormally slow kinetics, nonideal solution behavior resulting in “strange”-shaped traces, and high binding stoichiometries.49,52 One major disadvantage of SPR is the sequential nature of the sampling and the microfluid system. This makes the technique susceptible to compounds that precipitate air bubbles and anything that damages the surface, invalidating it for all subsequent injections. For this reason, it has become common practice to build SPR-compatible screening sets that are “prescreened” by SPR against a number of targets to remove aggregating, insoluble, covalent, and other problematic compounds. 52
The success of an SPR assay hinges on developing a protocol that immobilizes the protein in a highly functional state and finding suitable buffer conditions that maintain activity. In instances where no tools exist to validate the immobilized protein surface, knowledge of the protein integrity, stability, and general behavior is vital in maximizing the chances of success. For bromodomains, protein integrity can be conveniently checked by circular dichroism and NMR, whereas buffer compositions and DMSO tolerance can be assessed by methods such as DSF. The wealth of structural data is also able to guide the specific placement of immobilization tags and the design of inactive mutants as controls.
SPR was one of the techniques used to confirm the target of small-molecule activators of Apo-A1. 1 These compounds were initially identified with an unknown mechanism of action in a luciferase reporter assay but were found to pull down the tandem bromodomains of the BET family of proteins—namely, BRD2, BRD3, and BRD4—using a chemoproteomics approach. To characterize the molecules further, a number of methods (DSF, SPR, ITC, and X-ray) were employed to unequivocally demonstrate that their MOA was specific binding to each of the two bromodomains of the BET proteins at the Kac recognition site, with nanomolar affinities.
Single and tandem bromodomains of the BET family could be reproducibly coupled onto a CM5 chip by random amine coupling with high efficiency and good longevity, suggesting SPR could have been used for these proteins to screen compounds as well as for the detailed characterizations performed. The SPR affinities measured for the benzodiazepine BET inhibitor, GW841819X ( Fig. 3c ), to single and tandem bromodomains of BRD2, BRD3, and BRD4, were in the ten to low hundred nanomolar range and consistent with solution values by ITC and fluorescence. The sensitivity of current instrumentation suggests SPR can be used for fragment libraries and be feasible for screening bromodomains in the context of full or fuller length constructs.
Thermal shift differential scanning fluorimetry (DSF/ThermoFluor)
DSF or ThermoFluor monitors changes in protein thermal stability induced on ligand binding.53–55 This is visualized by the presence of a dye that binds more avidly to the denatured protein than to the native protein, resulting in increased fluorescence as the protein is temperature denatured. The baseline trace when no compound is present is useful for confirming protein integrity and can be used to find appropriate buffer conditions that optimize protein handling. 56 The degree of stabilization is not simply related to ligand affinity and is dependent on the thermodynamic nature of compound binding.57,58 Although it is difficult to generalize the limits of detectability without experimental observations on the system of interest, empirical observations suggest that typically compounds with low micromolar affinity are likely to be detectable in most instances. The protein consumption of DSF is somewhere between NMR and SPR, with typically 3 to 20 µL of 0.1 mg/mL protein used per well.
The major advantages of this technique are that it is plate based, is easily scalable, needs few bespoke reagents or equipment, and requires less expert knowledge to establish, run, and interpret compared to other LF methods such as NMR, SPR, and ITC. Like all LF methods, it is equally applicable to enzymes as well as to targets with no known function. 53 As a consequence of its broad utility and ease of use, it has been implemented in many laboratories as an accessible tool for hit identification and confirmation, often as a prelude to or in conjunction with other more quantitative methods.
A good example of its pan-target class potential has been demonstrated by the Structural Genomics Consortium (SGC), which employs a range of ligand-induced stabilization methods 59 and has used DSF to generate an interaction map for Ser/Thr kinases. 60 The potential to quickly establish an assay for a given protein is particularly attractive when establishing selectivity panels for new target classes such as bromodomains. Indeed, for the BET inhibitors ( Fig. 3c ) reported by GlaxoSmithKline (GSK) 1 (I-BET, GW84189X) and SGC 5 (+JQ1), both reported preliminary bromodomain selectivity assessments using this approach, among others. Notably, both also use ITC measurements to confirm their initial observations, acknowledging the limitations in quantitation by DSF, and preserve ITC as the gold-standard direct binding method.
Several features of DSF should be borne in mind when using this as a screening platform. DSF detects all interactions that result in an increase in protein thermal stability. Some of these can be nonspecific and of no functional relevance. The use of an active-site mutant would be one way of confirming specificity, as unlike NMR, there is no equivalent of a competition mode experiment. Another weakness is that DSF does not detect the ability of a compound to bind at ambient temperatures, but its ability to stabilize at temperatures where the protein begins to denature. Therefore, it is possible to have false-negative compounds that bind at room temperature but have strong temperature-dependent affinities. 61
It may be reasoned that compounds that show decreased affinity at elevated temperatures are less desirable in vivo tools because assays are normally performed at ambient temperatures, whereas mammals have body temperatures typically more than 10 °C above this. This, however, does not take into account that the assay protein is outside of its native protein and physiological environment, and the caveats associated with this may be particularly relevant for these small epigenetic reader domains, as the discussion of the temperature dependence for BRD2–BD1 in the section below highlights.
Use of isolated bromodomains
Techniques that are able to work with larger proteins rather than the minimal bromodomains such as those optimized for structural purposes may be beneficial. The binding properties of isolated bromodomains can be extremely construct dependent, even ignoring issues of multivalency. The binding of GW841819X to the N-terminal bromodomain of BRD2–BD1 illustrates this. 1 Binding of GW841819X to His6-tagged BRD–BD1 (67–200) was found to be highly temperature dependent, with an affinity of >1 µM at 26 °C and 46nM at 16 °C as determined by ITC. Yet in the context of a longer tandem domain construct, His-tagged BRD2–BD12 (1–473), binding was found to be in the tens of nanomolar range for the both BD1 and BD2 using the same technique. Mutational and kinetic studies suggest this is not a consequence of cooperative binding between the two BDs within the tandem constructs. Indeed, the C-terminal bromodomain of BRD2 exhibits no such temperature dependence, and similar affinities are observed for the isolated BD2 domain and to the same site in tandem proteins. The temperature dependence of this BRD2–BD1 domain is not due to inherent instability, as this construct shows all characteristics associated with a stable well-folded protein. It is highly expressed and could be purified to high yield (>10 mg/L) without difficulties. It could be concentrated beyond 10 mg/mL without need for high salt or glycerol to prevent aggregation, and it could be crystallized at room temperature to give apo crystals that diffracted to <2 Å. It also has a melting temperature of 52 °C, as determined by circular dichroism (CD). These observations serve to reinforce the difficulties in judging the suitability of isolated bromodomain reagents when the expected behavior and the affinities to binding partners are unknown. They also indicate the extreme care that should be taken to interpret in vitro selectivity data for these proteins.
Fluorescence Biochemical Formats
So far, this review has focused on LF methods that are particularly useful for identifying chemical starting points in a protein–protein system where little is known about the partner protein. Once suitable affinity peptides or ligands can be identified, many traditional biochemical formats become accessible. The biochemical methods that have been used to identify such assay tools and a number of reported fluorescence assays used for reader domains will now be discussed. As with experiences encountered with the use of LF approaches, special emphasis will be given to practical aspects that are likely to affect a range of formats such as intrinsic DMSO sensitivity and the weak and multivalent interactions that are a frequent feature of bromodomain–histone peptide interactions.
Identification of partner protein/peptide for assay development
Sequence-specific recognition within bromodomains is governed by the ZA and BC loops that exhibit high sequence and conformational variation ( Fig. 1b ). The few 3D peptide complex structures that exist show that peptides can occupy a large variety of channels along the surface defined by these loops, making it impossible to predict the likely binding mode of a given peptide or the cognate sequence for a given bromodomain. 62 Fortunately, the explosion of interest in epigenetic research has prompted an increasing collection of reagents to become available. Among these are histone peptide panels offered by several suppliers that contain an assortment of posttranslational modifications. These may be used to identify binding partners or substrates for epireader and epienzyme proteins, which form the basis for further assay development. A number of laboratories have also developed their own peptide profiling technologies.63,64 A major challenge in panning for peptide partners and their subsequent configuration into a robust assay for bromodomains is that most peptides reported have low affinities. Typical affinities are shown in Table 1 . These can be considerably lower than for other epireader domains and present a number of challenges. The use of high-sensitivity formats such as Alpha technology (AlphaLISA/AlphaScreen) can therefore be beneficial.
Selection of Peptide Affinities Reported for Bromodomains
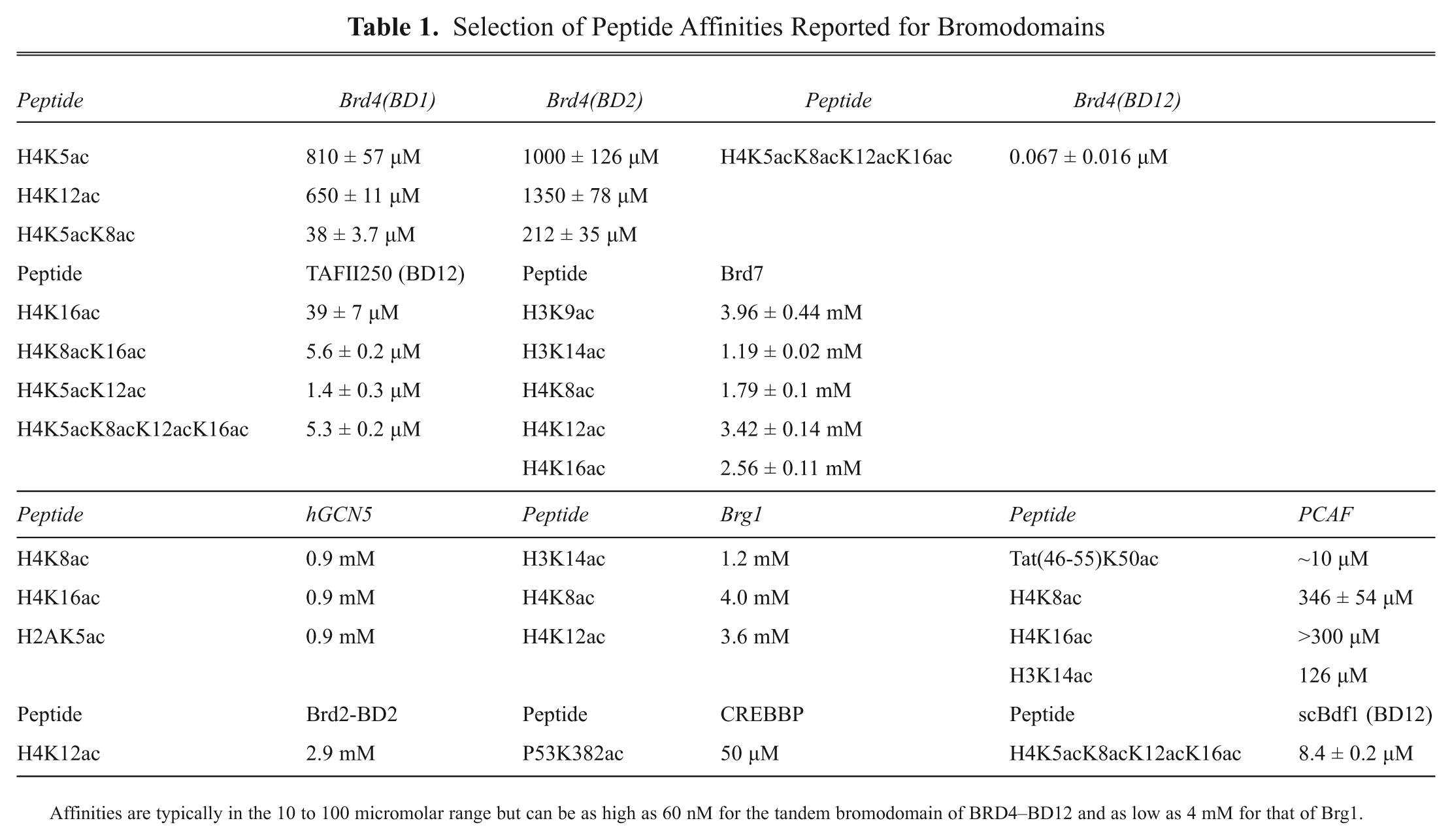
Affinities are typically in the 10 to 100 micromolar range but can be as high as 60 nM for the tandem bromodomain of BRD4–BD12 and as low as 4 mM for that of Brg1.
Alpha Technology for peptide identification and screening
Alpha Technology
The Alpha Technology (Amplified Luminescence Proximity Homogeneous Assay) is a homogeneous proximity-based reporting system.65,66 Laser excitation (680 nm) of phthalocyanine photosensitizer-containing donor beads excites ambient oxygen to give molecular oxygen with a single excited electron, known as singlet oxygen. This is captured by acceptor beads (incorporating thioxene, anthracene, and rubene or Europium derivatives) in close proximity (<200 nm) to generate a chemiluminescent signal in the 520- to 620-nm range. In the absence of an acceptor bead, the excited oxygen species relaxes to the ground state, and no signal is produced. The high sensitivity and low background of this assay technology make it especially suitable for detecting weak interactions 67 and enable compound screening without prohibitively large protein consumption. Application of this technology to epireaders, including four classes of methyl-lysine (Kme) reader domains7,8,67,68 (MBT, Chromo, PHD, and Tudor), has been published. As yet, the only documented bromodomain examples are that of BAZ2B, which has been screened at the NCGC (National Institutes of Health Chemical Genomics Center, http://www.ncgc.nih.gov/), and the use of BRD4–BD1, BRD4–BD2, and CREBBP AlphaScreen assays in the characterization of the BET inhibitor, JQ1 5 ( Fig. 3c ). A schematic of the assay configuration reported for BAZ2B is given in schematic form in Figure 4a . However, the conclusions of this screen are still to be published.
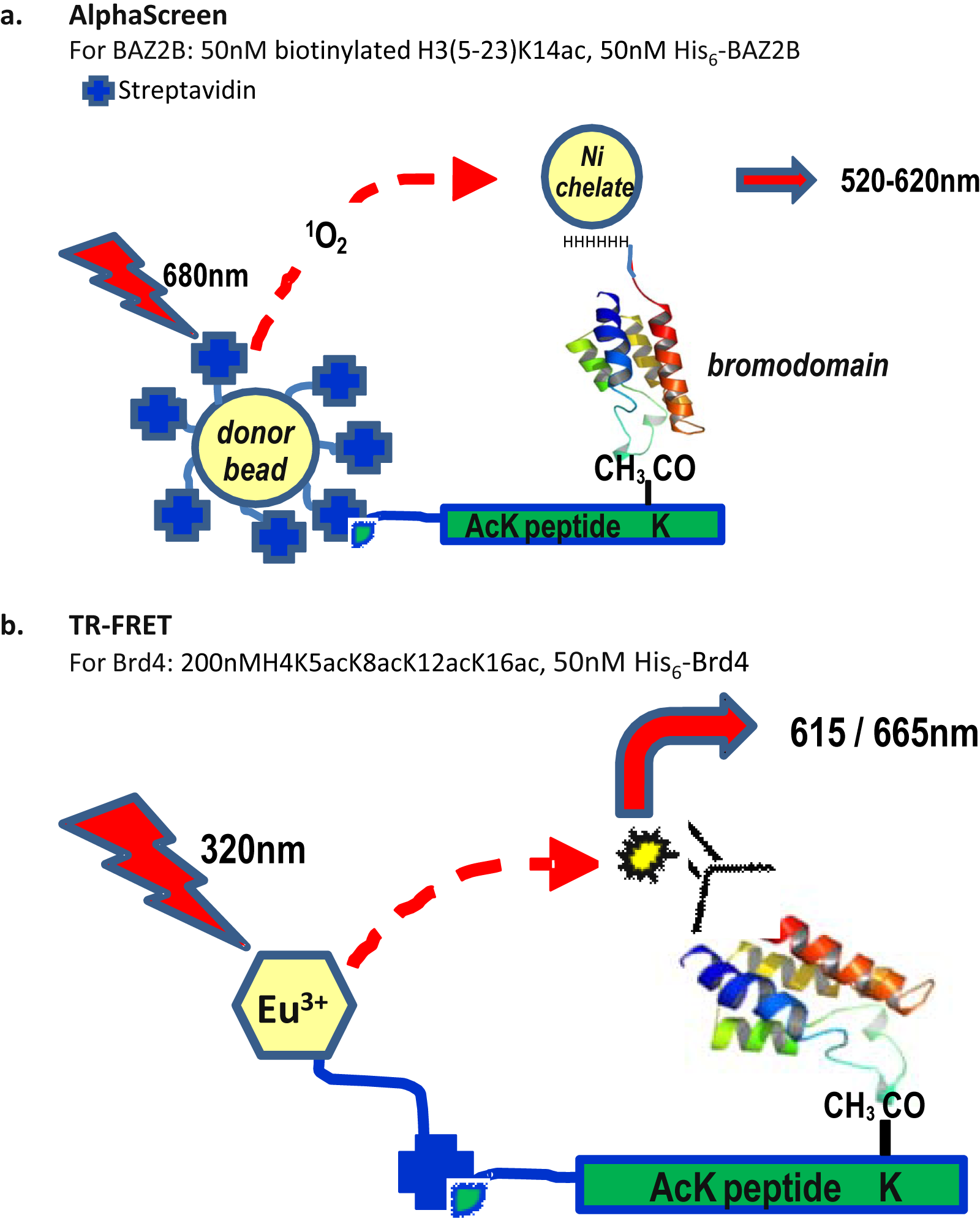
Schematic of AlphaScreen for BAZ2B and time-resolved fluorescence resonance energy transfer (TR-FRET) for BRD4. (
Experiences with methyl-lysine readers have considerable relevance to bromodomains, and therefore the literature for these targets and salient features that are likely to translate to bromodomains is highlighted.
Use for epireaders
Quinn et al. 67 demonstrated the broad utility of the AlphaScreen format to study the interactions of the three chromodomains, MPP8, HP1b, and CHD1; the tandem Tudor domain of JMJD2A; and the PHD domain of RAG2. All proteins were initially GST tagged to enable their coupling to the anti-GST-coated acceptor beads. Assays were then configured using a small number of biotinylated cognate trimethylated peptides (H3K9me3, H3K4me3, K4K20me3) immobilized onto the streptavidin donor beads. Competition experiments using corresponding nonbiotinylated peptides with the full range of methylated lysine states ((Kme0-3)) were used to confirm the preference for the trimethylated mark and also the specificity of the assay. As part of the optimization, an alternative His6-tagged MPP8 construct was explored with Ni-chelate acceptor beads. This gave superior signal to noise (S/N) as the Ni beads had higher binding capacity than their anti-GST counterparts, allowing a further reduction in reagent usage. Miniaturization to a 1536 format was also reported, and several of these targets have progressed into HTS within the NCGC. Their findings have yet to be fully described in the literature. Exploration of a number of tagging strategies may therefore be advantageous for the low bromodomain–peptide affinities expected.
Use of this technology has also been reported for the MBT class of epigenetic methylreaders.7,8,68 These possess several attributes similar to bromodomains. For example, MBTs are ~100 amino acid motifs found as multiple repeats within proteins. They are selective for mono- and dimethylated-lysine (Kme1/2), and like the Kac recognition within bromodomains, the methylated side chain is fully enclosed within a deep and narrow cavity, unlike the surface grooves of some other methyl-readers. The binding of MBT proteins to histone peptides appears to be less dependent on sequence specificity, and consequently peptide affinities are often lower than other methyl-readers, typically in the tens to hundred micromolar range, in line with those found for bromodomains.
L3MBTL1 is a prototypical MBT domain–containing protein that binds to H3K9Me with a reported activity of ~25 µM. 69 However, due to the avidity properties of a bead-based technology, 70 the multivalency of the interactions between a His6-tagged-L3MBTL1 fragment containing 3MBT repeats and the capture of a biotinylated H3K9Me peptide by tetravalent streptavidin, an apparent affinity of 54 to 88 nM was determined. Reassuringly, the IC50 of the non-biotinylated H3K9me peptide was 22 ± 3 µM, in good agreement with that previously measured by fluorescence anisotropy (FA). The exploitation of the effects of avidity may be one way to increase the sensitivity of weak interaction (e.g., by engineering a concatemer of the target reader domain).
L3MBTL1 was screened in a 384-well format (Z′ = 0.87) using the Sigma-Aldrich (St. Louis, MO) LOPAC(1280) library. This identified five hits, including two that had IC50 values less than 500 nM. Although these hits were reproducible in the AlphaScreen and showed the expected behavior in MOA analysis using this format (e.g., when the protein concentration was altered and when bovine serum albumin [BSA] or heat-inactivated His-L3MBTL1 was added), they were inactive in an FA histone-peptide-based assay. This highlights the utility of orthogonal confirmation assays. The same article indicated that this approach had also been applied to four other MBT proteins (SFMBT1, MBTD 1, L3MBTL3, and L3MBTL4), a Tudor domain protein (SGF 29), a PHD finger protein (PHF 13), and a chromodomain (CBX7) and that the Heightman group (Structural Genomics Consortium, Oxford, UK) had used it for bromodomains. This illustrates the broad potential applicability of the technology to reader domains, although the outcome of these additional efforts is still unpublished.
Recently, Herold et al. 7 demonstrated that a structure-based approach can be a viable and successful alternative to diversity screening for MBT domains. Using the co-crystal structure of H4K20Me2-bound L3MBT1, 71 the group adopted two complementary approaches. First, computational methods 8 were used to select a small set of 51 readily procurable compounds that were tested in dose–response mode in the AlphaScreen assays for L3MBTL1,3,4 and MBTD1, at a top concentration of 100 µM. Nineteen compounds demonstrated affinities of <100 µM against one or more of these MBT domains, giving an impressive hit rate of 37%. Second, a small number of peptidomimetics were synthesized based on the tripeptide (HFKme), where the Kme was replaced by a variety of simple diamines. This again identified a number of active compounds. A counterscreen using a biotin-His6-peptide in the absence of protein 68 had revealed that a subset of the small-molecule scaffolds showed assay interference. Therefore, hits from the AlphaScreen were validated using ITC, which also confirmed a 1:1 stoichimetry of interaction. To further corroborate their binding mode, ITC was used to show that an active-site mutant was no longer able to bind these compounds. The final validation was the co-crystal structure of a 5-µM optimized compound within the expected recognition site of L3MBT1.
The methyl-reader examples above illustrate the potential for the Alpha format but also reinforce the need for an interference counterscreen and the use of orthogonal methods for hit confirmation. ITC and X-ray crystallography seem to have been the definitive methods of choice for epireaders thus far.
Effect of buffer components and consideration for peptide scanning
It is well known that buffer choice is very important for the Alpha technology. The wrong buffer choice can result in severe reduction or even elimination of the signal window. In the L3MBTL1 example, although a signal could be observed in an initial buffer of 20 mM Tris (pH 8), 2 mM dithiothreitol (DTT), and 0.1% BSA, the high background due to nonspecific binding could be improved by replacing the BSA with 25 mM NaCl and 0.05% Tween. Clearly, such optimization is more difficult when probing for new peptide partners, so if possible, a number of buffers should be tried.
Effect of DMSO on L3MBT1 and bromodomains
The L3MBT1 AlphaScreen assay was reported to be very sensitive to DMSO. This sensitivity was not unique to the AlphaScreen format but seemed to be a particular attribution of the protein, as DMSO sensitivity was also reported in the FA assay, 69 and the presence of DMSO completely abolished the ability to detect binding in the ITC. Consequently, all titrations were conducted in aqueous media with the ligand being dissolved directly into ITC buffer.
DMSO has been shown to be a direct competitor for the Kac site for bromodomains. DMSO is therefore likely to be an important consideration for bromodomain assays, although it is expected that the precise DMSO sensitivity will be protein and assay format dependent.
Reported cases of using the traditional fluorescence assay
Traditional fluorescence formats (e.g., fluorescence anisotropy and TR-FRET) have been used for bromodomains where suitable ligands of appropriate affinity have been identified. These are well-established screening formats, so their implementation and results are reviewed only briefly below, with prominence given to themes that may have broad relevance.
A number of laboratories have reported using an FA format to monitor bromodomain–peptide binding. This includes a number of studies on human PB1 (polybromo-1).72,73 PB1 protein is thought to localize the Polybromo, BRG1-associated factors chromatin–remodeling complex to kinetochores during mitosis via direct interaction of its six bromodomains with acetylated nucleosomes. Using His-tagged single-bromodomain constructs of this protein and the fluorescein-labeled H3 peptides listed in Table 2 , the Michigan group determined the binding specificities by steady-state fluorescence anisotropy. The results indicate that the single bromodomains bind specific Kac sites with submicromolar affinity ( Table 2 ). This group subsequently evaluated the kinetics 74 and thermodynamics 75 of binding using time-resolved fluorescence and by applying van’t Hoff analysis to the temperature dependence of binding. Recently, this work was complemented by that of the group of Ming-Ming Zhou, who used NMR to elucidate the structural basis for the interaction of H3K14Ac with PB1–BD2. However, the affinities they determined by NMR were far higher for this interaction (~500 µM) 76 than determined previously by FA (1.4 µM). 40 Interestingly, this group also found a seemingly different profile of peptide preferences to that previously determined. For example, their ELISA-based format did not detect interactions of PB1–BD2 with H3K9ac or H3K18ac but did detect an interaction with H3K36ac, which was absent from the FA study. This example highlights one of many where differential histone peptide profiles have been reported for the same protein. There may be numerous reasons for this. In this comparative example, there were differences in assay format, peptide length, range of peptide sequences profiled, construct length for each domain, and the use of different tagging strategies for the proteins ( Table 2 ). All of these may contribute to observed variations and emphasize some of the difficulties in translating peptide profiling results between formats. The low affinities of peptides for bromodomains are likely to hinder matters as the differential between specific and nonspecific binding may be modest.
Peptide Affinities for the Six Bromodomains of Human Polybromodomain–1 Determined in a Fluorescence Anisotropy Assay with the Fluorescein Peptides and His-Tagged Constructs
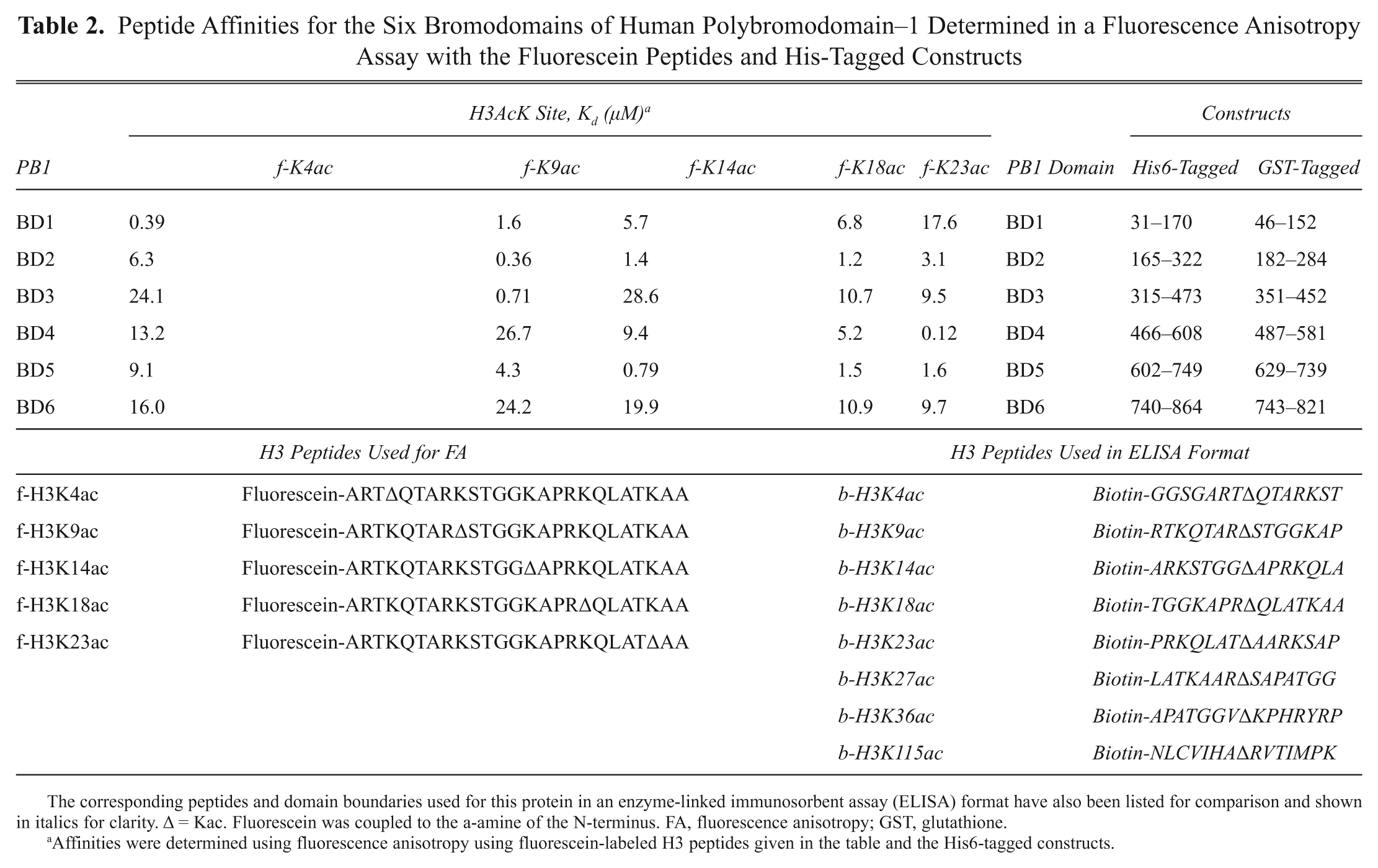
The corresponding peptides and domain boundaries used for this protein in an enzyme-linked immunosorbent assay (ELISA) format have also been listed for comparison and shown in italics for clarity. Δ = Kac. Fluorescein was coupled to the a-amine of the N-terminus. FA, fluorescence anisotropy; GST, glutathione.
Affinities were determined using fluorescence anisotropy using fluorescein-labeled H3 peptides given in the table and the His6-tagged constructs.
Fluorescent assays based on small-molecule probes have also been reported recently. In a search for a CREBBP inhibitor, cyclized peptides based on p53-K382ac were characterized by NMR and a fluorescence assay. 2 The FA probe used was a fluorescein derivative of an analog of Ischemin, which had been previously discovered by NMR screening ( Fig. 5b ). The reported affinity of the probe was 75 µM. Following the identification of a higher affinity cyclic peptide from this effort, a fluorescein version of the peptide was made ( Fig. 5c ). This may provide an improved assay tool for CREBBP.
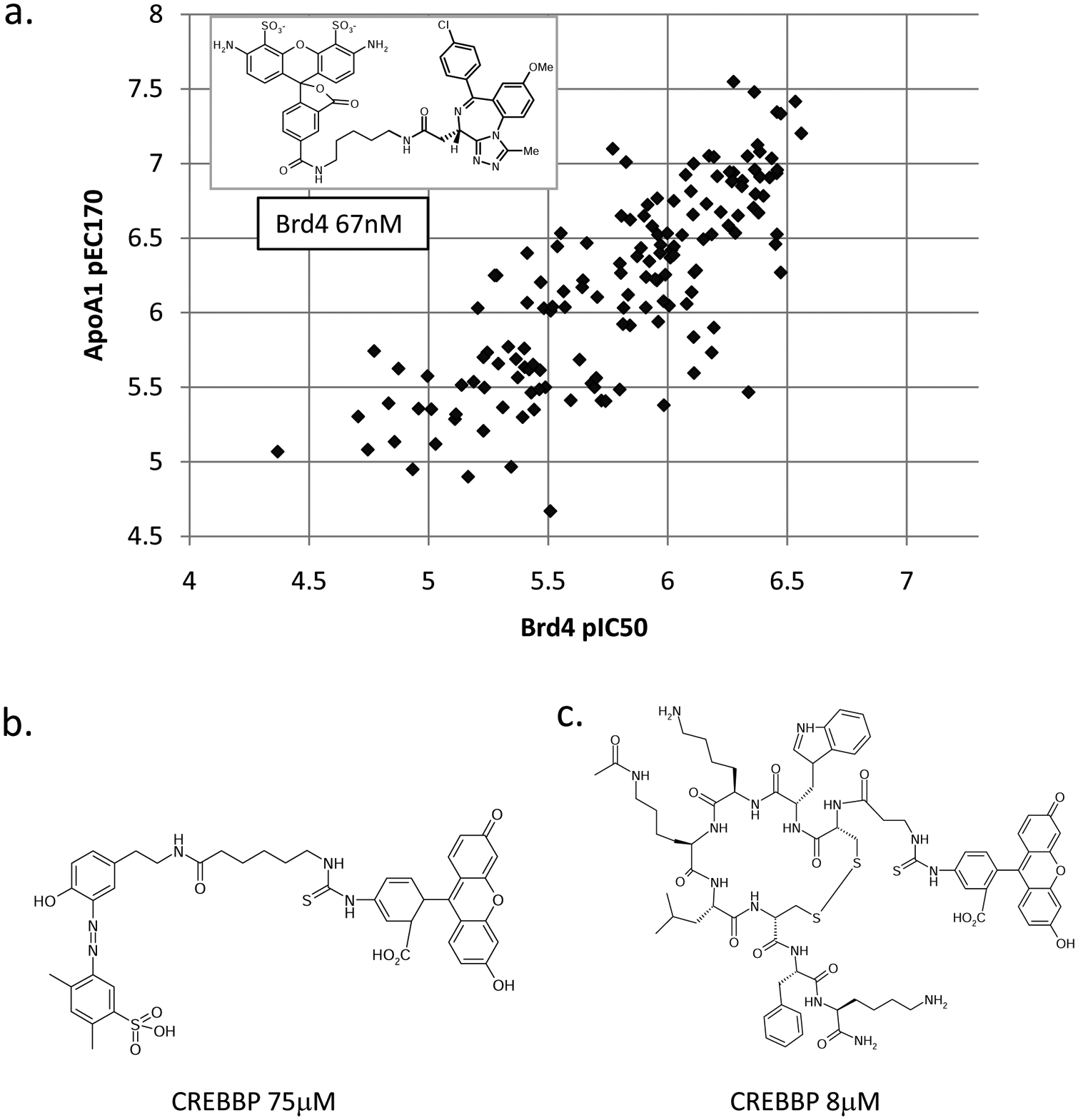
Fluorescent bromodomain probes and correlation of BRD4 fluorescence anisotropy (FA) data with cellular Apo-A1 upregulation. (
In a similar scenario, a potent FA probe based on the nanomolar benzodiazepene compounds reported as BET inhibitors has also been demonstrated ( Fig. 5a ). The profiling of 150 benzodiazepene compounds using this assay showed an excellent correlation between in vitro binding to BRD4 and upregulation of Apo-A1 in a luciferase reporter readout. A TR-FRET histone displacement format ( Fig. 4b ) was also reported for BRD4 within the same publication to directly confirm the ability of these compounds to inhibit peptide binding.
In all these instances, knowledge of the ligand structure–activity relationship (SAR) and its binding mode within the protein faciliated placement of the fluorescent label. The relatively open entrance to the Kac site of BRDs generally makes the conversion of inhibitors to fluorescent tools straightforward if the chemistry of attachment is amenable.
Cellular approaches
An important area of epigenetic drug discovery beyond the scope of this review is the use of cell-based assays for hit identification and mechanistic deconvolution.6,11,77–79 For example, phenotypic readouts measuring Apo-A1 upregulation and anti-inflammatory responses were responsible for the initial identification of the benzodiazepine 22 and thienodiazepene80–82 templates for BET shown in Figure 3c . Screening compounds for inhibitors of the BRD2–H4K12ac interaction using an intracellular FRET format also have been recently reported and have successfully identified a benzimidazole compound confirmed to bind to BRD2 by crystallography. 6
Conclusions
The therapeutic potential of epigenetic reader domains represents an unexploited opportunity for small-molecule drug intervention. The emerging armory of techniques, ranging from label-free approaches such as NMR, SPR, and ITC to Alpha technologies and traditional fluorescent formats, provides the tools to design an efficient hit identification and triaging strategy for these domains that can be tailored to the target of interest.
Footnotes
Acknowledgements
Thanks to Paul Bamborough, Gareth Wayne, Daniel Gaughan, Onkar Singh, and Mike Hann for helpful comments and careful proofreading.