
Abstract
H+,K+-ATPase is a key enzyme in the process of gastric acid secretion, and proton pump inhibitors (PPIs) have been accepted as one of the most effective treatments for peptic ulcer and gastroesophageal reflux disease. To discover a novel class of PPIs, the authors screened a low-molecular-weight compound library and identified two prospective acid blockers that were pyrrole derivatives. Both compounds inhibited H+,K+-ATPase in a reversible and potassium-competitive manner. These compounds led to the development of TAK-438 (1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine monofumarate), which is currently undergoing clinical trials as a novel potassium-competitive acid blocker for the treatment of acid-related diseases.
Keywords
Introduction
Gastric H+,K+-ATPase belongs to a class of ion translocating P-type ATPases.1–3 Other enzymes in this class include Na+,K+-ATPase and Ca+-ATPases. Gastric H+,K+-ATPase is responsible for the final step of gastric acid secretion. Proton pump inhibitors (PPIs) are drugs targeting gastric H+,K+-ATPase, which are widely used for the treatment of gastroesophageal reflux disease, peptic ulcer, and other acid-related diseases. 4 The PPIs currently in use are substituted pyridylmethylsulfinyl benzimidazole prodrugs. They accumulate in the acidic secretory canaliculi of gastric parietal cells and then undergo acid-catalyzed chemical rearrangement to yield an active thiophilic species (a sulfenic acid or sulfonamide) that binds to various cysteines accessible from the luminal surface of gastric H+,K+-ATPase, thus forming disulfides.5–7
Despite PPIs’ well-documented efficacy and safety, several areas where treatment could be further refined or enhanced have been suggested. PPIs have a relatively slow onset of pharmacological action and may require 3 and 5 days of dosing to achieve maximum acid suppression and symptom relief. 8 PPIs may also fail to provide 24-h suppression of gastric acid, and nighttime acid breakthrough can occur even with twice-daily dosing.9,10 PPI therapy is insufficient for the pH control needed for preventing rebleeding after endoscopic hemostasis of patients with gastrointestinal bleeding.11,12 Moreover, because the hepatic metabolism of PPIs is dependent on the CYP2C19 system, they are affected by the CYP2C19 genetic polymorphism and may have potential for clinically significant pharmacokinetic drug interactions such as those with clopidogrel. 13
To discover novel compounds with alternative mechanisms of H+,K+-ATPase inhibition and the potential to overcome such limitations of PPIs, we focused on K+ competition. Because K+ is required for dephosphorylation of this enzyme and subsequent conformational changes, 14 blocking the access of K+ is an effective method of inhibition.15–17
We screened a low-molecular-weight compound library by using a high-throughput assay system to indentify H+,K+-ATPase inhibitors. Then we investigated the profile of the compounds detected by primary screening. As a result, two compounds were chosen for further study. Both compounds 1 and 2 inhibited H+,K+-ATPase in a concentration-dependent manner, with IC50 values of 0.48 µM and 0.44 µM, respectively. Both compounds also showed reversible and K+-competitive inhibition. And these compounds led to the development of TAK-438. 18
Materials and Methods
Materials
Lansoprazole was synthesized by Takeda Pharmaceutical Company Limited (Osaka, Japan). Canine kidney Na+,K+-ATPase, ATP, and SCH28080 were purchased from Sigma-Aldrich (St. Louis, MO).
Preparation of porcine gastric H+,K+-ATPase
Porcine stomachs were obtained from Katayama Chemical, Ltd. (Osaka, Japan). The fundus of each stomach was isolated, rinsed with tap water, and washed with 3M NaCl to remove superficial cells, cell debris, and mucus. Then the oxyntic cell-rich mucosa was scraped off and homogenized in a homogenizing buffer (0.25M sucrose, 1 mmol/L EDTA, and 10 mmol/L Tris-HCl, pH 6.8) with a Polytron homogenizer (Kinematica, Bohemia, NY). The homogenate was centrifuged at 20 000 g for 30 min, and the supernatant was further centrifuged at 100 000 g for 90 min. The pellet was resuspended in homogenizing buffer, after which it was layered over 7.5% (w/w) Ficoll in the homogenizing buffer and centrifuged at 100 000 g for 60 min. The microsomal fraction (concentrated in the middle layer) was collected, diluted with homogenizing buffer, and centrifuged again at 100 000 g for 90 min. Then the pellet was collected and suspended in homogenizing buffer at a final protein concentration of 0.5 mg/mL, after which the suspension was stored at –80°C until its protein content was determined with a Quick Start Bradford protein assay kit (Bio-Rad, Hercules, CA).
Measurement of H+,K+-ATPase activity
Gastric H+,K+-ATPase activity was measured by quantifying the release of inorganic phosphate from ATP 19 in a 384-well format. Reactions were performed in a reaction mixture of 50 µL containing 2.5 mg/L vesicles, 50mM Hepes-Tris (pH 6.5 or 7.4), 5 mM MgCl2, 8 mM KCl, 10 μM valinomycin in the presence of the test compound or vehicle. After preincubation at 37 °C for 30 min, the reaction was initiated by the addition of ATP at a final concentration of 0.2 mM and incubation at 37 °C for 20 min. The reaction was stopped by adding 15 µL of dye reagent containing 0.1% w/v malachite green, 1.5% w/v hexaammonium molybdate, and 0.2% v/v Tween 20 (Sigma-Aldrich, St. Louis, MO) in 4 N H2SO4, after which the absorbance was measured at 620 nm with a Wallac HTS (PerkinElmer, Waltham, MA). K+-dependent ATPase activity was calculated as the difference between the activity in the presence and absence of KCl. For the controls with 0% inhibition and 100% inhibition, enzymatic reactions were carried out in the presence of 1% DMSO and 10 µM SCH28080, respectively.
Measurement of Na+,K+-ATPase activity
The activity of canine kidney Na+,K+-ATPase (Sigma) was measured in a 50 µL reaction mixture containing 0.32 µg of protein, 50 mM Hepes-Tris (pH 7.4), 2 mM MgCl2, 100 mM NaCl, and 10 mM KCl with the test compound or vehicle. After preincubation at 37 °C for 30 min, the reaction was initiated by addition of ATP at a final concentration of 0.1 mM and incubation at 37 °C for 25 min. Release of inorganic phosphate was measured in the same way as for the H+,K+-ATPase assay, except that reactions were carried out in the presence of 100 µM ouabain as the 100% inhibition control.
Reversibility test
Reversibility was investigated by dilution inhibition experiments, with the recovery of H+,K+-ATPase activity being measured after dilution of the incubation mixture. The starting concentration of each compound was 20-fold higher than its IC50, and 200-fold dilutions were done at a constant KCl concentration (25 mM). H+,K+-ATPase activity was set at 100% by measuring it at each dilution without an inhibitor.
Affinity selection mass spectrometry (ASMS)
Binding of the test compounds to H+,K+-ATPase was investigated by ASMS20–22 using porcine gastric vesicles as a source of H+,K+-ATPase.
These vesicles were initially incubated with a competitor compound in binding buffer (50mM Hepes-Tris [pH 6.5], 5 mM MgCl2, 8 mM KCl, 10 μM valinomycin, and 0.2 mM ATP) at room temperature for 30 min. To determine nonspecific binding, a compound that showed competitive binding with the ligand was used as the competitor. Then a test compound was added and incubation was done for 60 min. After that, protein-ligand complexes were separated from the unbound compound by rapid low temperature (<30 s at 4 °C) size-exclusion chromatography. Subsequently, formic acid (0.13%) was added to promote dissociation of ligands from the protein. The dissociated ligands were applied to a C18 high-performance liquid chromatography column, followed by an electrospray-ionization mass spectrometer (API5000; Applied Biosystems, Foster City, CA) for identification and quantification.
Results
HTS results
The high-throughput screening was completed on 560000 compounds using the colorimetric assay with porcine gastric vesicles at a single concentration of 2 µM.
The average signal/background ratio and Z′ factor values were 2.8 and 0.74, respectively. During initial screening, 96 compounds showed >50% inhibition at 2 µM and were selected for further assessment of activity.
Profiling of selected compounds
Of the above-mentioned 96 compounds, 34 compounds showed dose-dependent inhibitory activity. We conducted further profiling assays of these 34 compounds to assess the following features: (1) selectivity, (2) reversibility of inhibition, (3) substrate competition, and (4) effect of pH on the inhibitory activity.
As a result of these further characterization procedures, two compounds (1 and 2; Fig. 1 ) showed a promising profile.
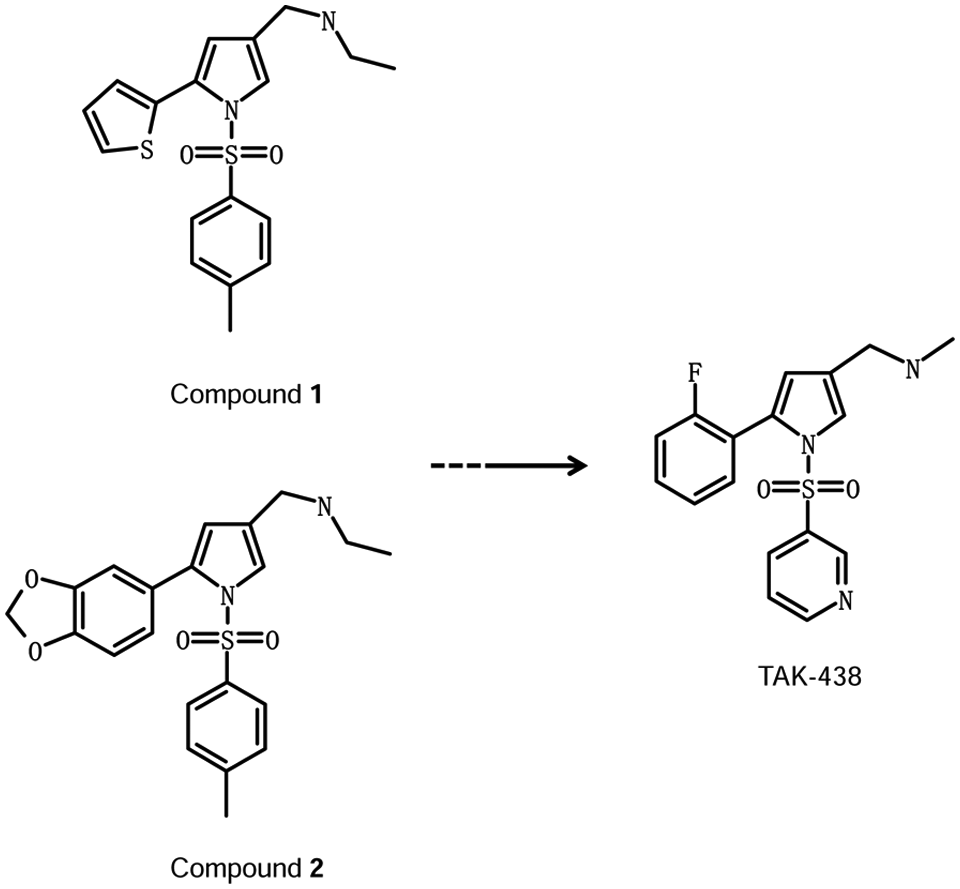
Structures of 1, 2, and TAK-438.
Selectivity
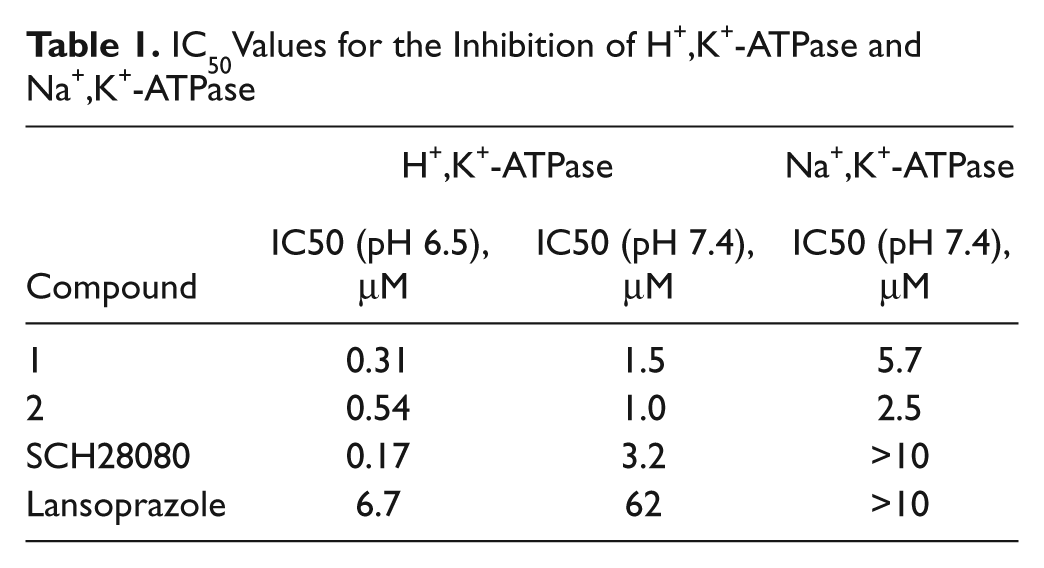
To assess selectivity for H+,K+-ATPase, we screened the activity of these compounds against Na+,K+-ATPase. 1 and 2 inhibited H+,K+-ATPase activity with IC50 values of 0.31 µM and 0.54 µM, respectively, and both were about 5 to 10 times more potent at inhibiting H+,K+-ATPase than Na+,K+-ATPase ( Table 1 ).
IC50Values for the Inhibition of H+,K+-ATPase and Na+,K+-ATPase
Reversibility of inhibition
After dilution, each compound concentration changed from 20-fold IC50 to 0.1-fold IC50. According to the ideal concentration-response equation, these concentrations correspond to approximately 95% and 9% inhibition. The inhibition of SCH28080, compound 1, and compound 2 were significantly attenuated by dilution, in agreement with that theoretical value. In contrast, the irreversible inhibitor lansoprazole displayed no recovery of ATPase activity ( Fig. 2 ).
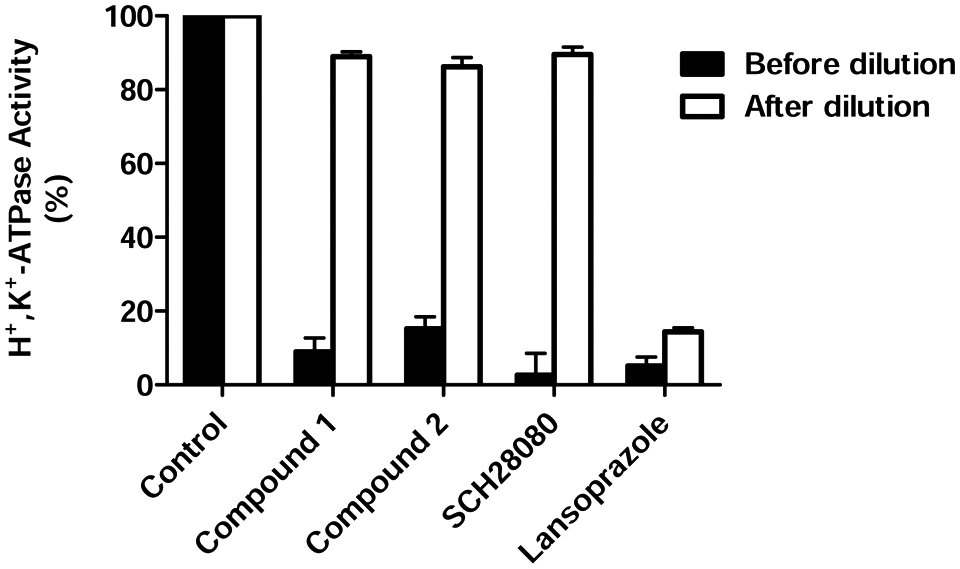
Reversibility of H+,K+-ATPase inhibition. The effect of 1 (10 µM), 2 (10 µM), lansoprazole (20 µM), and SCH-28080 (3 µM) on H+,K+-ATPase activity was measured after (open columns) and before (closed columns) 200-fold dilution. Results are expressed as the mean ± SE (n = 6).
These data indicated that inhibition of H+,K+-ATPase by 1 and 2 was reversible.
Substrate competition
To explore the mechanisms of action of the hit compounds, the dependence of the enzyme activity on potassium concentrations at several inhibitor concentrations was investigated. A global fitting to the equation for models of enzyme inhibition was performed using GraphPad (GraphPad Software, Inc, La Jolla, CA) nonlinear least-squares analysis. The inhibition by two compounds as well as SCH28080 was best described as competitive ( Fig. 3 ).
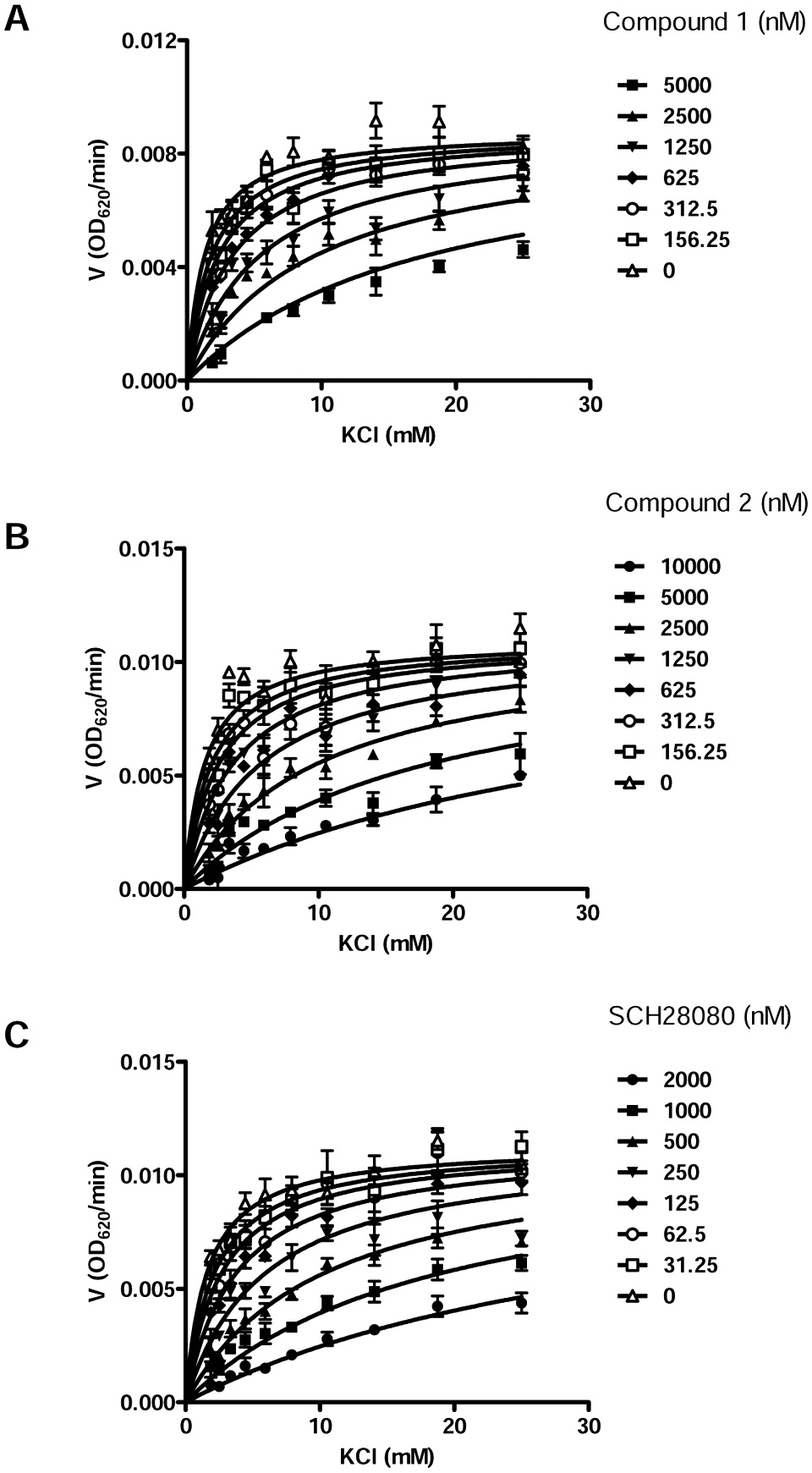
Determination of inhibition mechanism. Global fitting of velocity versus potassium concentration data in absence or presence of 1 (
Effect of pH
The inhibitory activity of 1 and 2 was slightly stronger at pH 6.5 than at pH 7.4 ( Table 1 ).
ASMS
When we conducted ASMS binding experiments using porcine gastric vesicles as a source of H+,K+-ATPase,
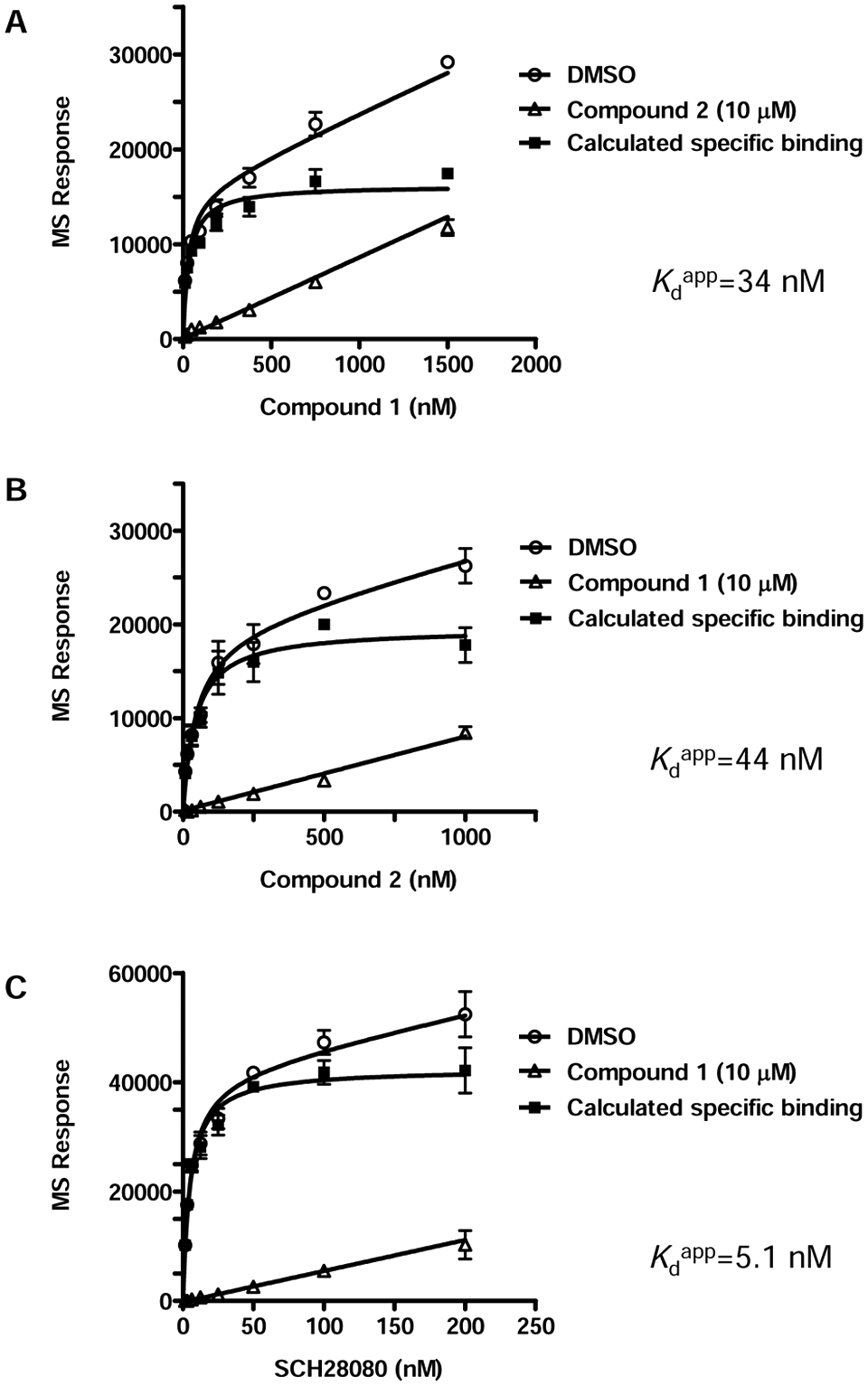
H+,K+-ATPase binding affinity of 1 (
The effect of lansoprazole was also studied. Lansoprazole was added to porcine gastric vesicles and preincubated at room temperature for 30 min. Then 1 or 2 was added, and the mixture was incubated for 60 min and subjected to ASMS analysis. It was found that pretreatment with lansoprazole reduced the binding of both compounds ( Fig. 5 ).
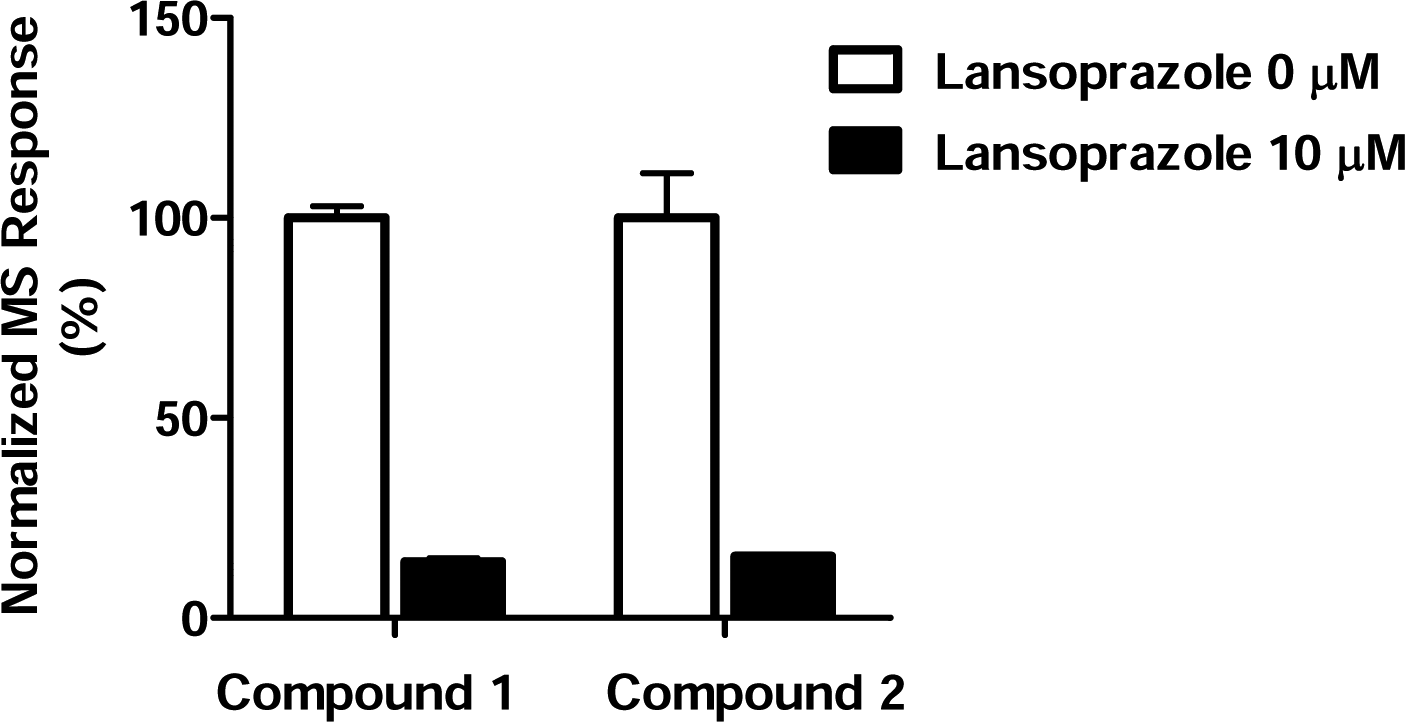
Effect of lansoprazole on binding of 1 and 2 in affinity selection mass spectrometry (ASMS) binding experiments. Lansoprazole was added to porcine gastric vesicles and preincubated at room temperature for 30 min. Then 1 or 2 was added, and the mixture was incubated for 60 min before ASMS analysis.
Discussion
The PPIs currently in clinical use are substituted pyridylmethylsulfinyl benzimidazole prodrugs that undergo acid-catalyzed chemical rearrangement in vivo to yield an active thiophilic species (a sulfenic acid or sulfonamide), which binds to various cysteines accessible from the luminal surface of gastric H+,K+-ATPase to form disulfides. This mechanism of gastric acid inhibition by PPIs is related to their main drawback, which is a requirement for acid secretion into the secretory canaliculi of gastric parietal cells to promote accumulation of the prodrug and conversion to its active form. 23
Here we report the discovery of two novel H+,K+-ATPase inhibitors, 1 and 2, which inhibit H+,K+-ATPase activity in a reversible and potassium-competitive manner. To obtain promising H+,K+-ATPase inhibitors, we developed a high-throughput screening protocol for evaluating gastric H+,K+-ATPase activity by the malachite green–based assay and then screened a low-molecular-weight compound library.
We investigated the effect of pH by studying the inhibition of enzyme activity at pH 6.5 and pH 7.4. As a result, the two compounds were less influenced by pH than SCH28080 or lansoprazole.
A previous report 15 suggested that the active species of SCH28080 was its protonated form. The pKa of SCH28080 is 5.5, and its inhibition is stronger at pH 6.4 compared with that at pH 7.3, whereas a permanent cation analogue of SCH28080 shows little difference in potency between pH 6.4 and pH 7.3. In contrast, both 1 and 2 have calculated pKa values of 9.3, so it can be assumed that they exist as the protonated form in neutral solutions as well as in acidic solutions. Therefore, it is unlikely that protonation was related to the effect of pH on the potency of 1 and 2. Further investigations will be needed to assess the role of other possible factors, such as protonation of H+,K+-ATPase amino acid side chains.
In ASMS experiments, we used porcine gastric vesicles and obtained saturable one-site binding curves. Competitive binding was observed for 1, 2, and SCH28080. These results suggest that all three compounds bind to the same site or nearby sites.
Moreover, apparent Kd values of 34, 44, and 5.1 nM were obtained for 1, 2, and SCH28080, respectively. However, these values were nearly one-tenth of their respective IC50 in the enzyme assay.
P-type ATPases are characterized by an ATP hydrolysis coupled ion transport cycle that proceeds through several intermediates. Moreover, each intermediate state has distinct affinity for ATP and ion ligands. 4 Because we expected the same intermediates could present, we used the ASMS binding buffer that had the same composition as the enzyme assay. Therefore, there was no difference in binding conditions that cause this discrepancy of affinities.
To separate protein-ligand complexes from unbound compounds, size-exclusion chromatography was conducted in the ASMS experiments. In the size-exclusion chromatography step using Millipore’s MultiScreen-HA filter plate packed with Sephadex-G50 gel, (GE Healthcare, Piscataway, NJ) protein recovery was <30% (data not shown). Because the source of H+,K+-ATPase used in this study was vesicle, adsorption is considered to be one of reasons of low recovery. However, it is unclear that this step is a cause of the discrepancy between Kd and IC50. Although our ASMS experiments certainly showed specific binding of the test compounds to H+,K+-ATPase, the cause of higher binding affinity compared with enzyme assay requires further investigation.
We also investigated the effect of lansoprazole by ASMS. Lansoprazole is transformed by acid-catalyzed conversion into a tetracyclic cationic sulfenamide that inhibits H+,K+-ATPase activity by reacting with SH groups of the enzyme. 24 For our ASMS method, the binding buffer was adjusted to pH 6.5, which would cause acidification of lansoprazole to some degree. It should also be noted that ATP is contained in the binding buffer, because it has been reported that ATP stimulates accumulation of H+ in vesicles by H+,K+-ATPase-mediated H+ transport. 25 These changes would be expected to promote the binding of lansoprazole to H+,K+-ATPase. In fact, we found that preincubation with lansoprazole interfered with the binding of 1 and 2. This result was consistent with a previous report that the inhibitor binding sites for benzimidazoles and SCH28080 overlap, because mutation of Cys-813 has an effect on affinity for SCH28080. 26
In summary, we conducted high-throughput screening of a low-molecular-weight compound library and identified two novel H+,K+-ATPase inhibitors (1 and 2). Both of these compounds inhibited H+,K+-ATPase in a reversible and potassium-competitive manner. Further investigation of these compounds led to the development of TAK-438, which is currently undergoing clinical trials as a novel potassium-competitive acid blocker for the treatment of acid-related diseases.
Footnotes
Acknowledgements
We thank Dr. Hideaki Nagaya for helpful suggestions.