
Abstract
1-deoxy-D-xylulose 5-phosphate reductoisomerase (Dxr) is involved in the synthesis of isoprenoids by the methylerythritol phosphate pathway. Dxr is essential in Mycobacterium tuberculosis (Mtu), absent in humans and amenable to structure-aided design. To further assess the druggability of the enzyme, the energetics of binding of fosmidomycin to Mtu Dxr was studied by isothermal calorimetry. Binding was enhanced by nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) and driven by enthalpy (ΔH –10.2 kcal/mol, ΔS 1.1 cal mol−1K−1). This suggests the possibility of finding novel inhibitors that bind enthalpically, making Dxr an attractive target. The cost of the Dxr substrate, 1-deoxy-D-xylulose-5-phosphate, for high-throughput screening (HTS) is prohibitive. Hence, an HTS assay that couples Dxr to the upstream enzyme 1-deoxy-D-xylulose-5-phosphate synthase (Dxs), also a valid target, was developed. A high concentration of NADPH was used to bias it to detect Dxr inhibitors that bind like fosmidomycin. The assay Z′ was 0.75. It was equally sensitive to inhibitors of Dxs and Dxr, that is, fosmidomycin and fluropyruvate inhibited it with IC50s similar to that in the individual enzyme assays (79 vs 54 nM for fosmidomycin). To distinguish inhibitors of Dxs from Dxr, individual enzyme assays and a microplate thermofluor binding assay were developed. The assay simultaneously screens two targets and is cost-effective.
Keywords
A
Isoprenoids play an important role in building the bacterial cell wall. The lipid carriers essential for the synthesis of the mycolic acid–arabinogalactan–peptidoglycan complex of the cell wall of Mycobacterium tuberculosis (Mtu) are isoprenoids. They are also involved in key metabolic functions such as the electron transport system (e.g., ubiquinone and menaquinone). Thus, inhibition of enzymes involved in synthesis of isoprenoids should bring about a pleiotropic effect.
Isoprenoids are synthesized using isopentenyl pyrophosphate and dimethylallyl pyrophosphate as building blocks. These building blocks are made by either of two pathways: a mevalonate pathway in humans and a methylerythritol phosphate (MEP) pathway in Mtu and most bacteria. The MEP pathway, which is often referred to as the nonmevalonate pathway, is absent in Homo sapiens. 2 The pathway enzymes are essential for the survival of Escherichia coli. Thus, the MEP pathway presents a host of valuable targets for the discovery of novel antibacterial or antimycobacterial agents. Here, the first two enzymes of the MEP pathway, 1-deoxy-D-xylulose-5-phosphate synthase (Dxs) and 1-deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr) are addressed. Dxs catalyzes the synthesis of 1-deoxy-D-xylulose-5-phosphate (DXP) from glyceraldehyde-3-phosphate (G-3P) and pyruvate in the first committed step of the MEP pathway. Dxr converts DXP to MEP concomitant with oxidation of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) to nicotinamide adenine dinucleotide phosphate (NADP) in a metal ion-dependent reaction ( Fig. 1 ).
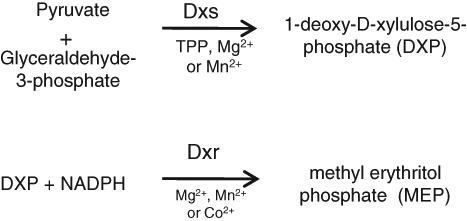
Schematic of enzyme reactions catalysed by 1-deoxy-D-xylulose-5-phosphate synthase (Dxs) and 1-deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr).
Dxr is essential for survival of Mtu 3 (and in-house data). Fosmidomycin is a potent inhibitor of Dxr, and crystal structures of Mtu Dxr in complex with NADPH and fosmidomycin have been solved, 4,5 enabling structure-aided design. Fosmidomycin inhibits Mtu Dxr, but it does not inhibit the growth of the bacterium, presumably because it does not penetrate the cell or is effluxed out. 6 However, fosmidomycin can cure mice of a bacterial infection 7 and has even undergone clinical trials 8 for malaria and bacterial infections, suggesting that novel inhibitors of Dxr have the potential to become antibacterial drugs.
The intention was to use high-throughput screening (HTS) of a compound library to find novel inhibitors of Dxr as chemical starting points for drug discovery. However, many antibacterial targets have been screened with little success. 9 Given the time and cost involved in an HTS, in our process, typically target enzymes are evaluated against several criteria, including druggability, prior to developing an assay for HTS. Prime among these criteria is that the enzyme is essential for the survival of the bacterium and that no human homolog exists. Having a crystal structure, especially that with an inhibitor bound, or a substrate, is an added advantage. Although the interpretation of the word druggability varies widely, one of the aspects considered by us is whether an inhibitor is known for the target, suggesting to us that the enzyme has a pocket that is accessible to and suitable for the binding of a small molecule. The other is the energetics of binding of the inhibitor.
Intuitively, it would seem that inhibitors that bind primarily with an enthalpic component are more specific to the target 10 and have fewer off-target effects than those where the binding is driven by entropy. Also, data from HIV-1 protease inhibitors and statins suggest that best-in-class compounds have a much enhanced enthalpic binding component compared to first-in-class compounds. 11 In our view, a protein that has an inhibitor that binds primarily via enthalpy, rather than entropy, is more druggable. It suggests to us that the binding pocket has the ability to make these interactions, and thus the chance of finding novel inhibitors that primarily bind with an enthalpic component is reasonably good. Therefore, the energetics of binding of fosmidomycin to Dxr was studied by isothermal calorimetry (ITC). Although inhibition of both E. coli 12 and Mtu Dxr by fosmidomycin has been reported, 6 there is no report on the energetics of binding of fosmidomycin to Dxr. ITC measures the enthalpy, the stoichiometry of binding, and the affinity of the ligand for the protein. With this information, the entropy of binding can be calculated.
Having decided to use HTS to find inhibitors of Dxr, a compatible assay was needed. An HTS assay has been reported for Dxr, but it is a binding assay that would detect only inhibitors that compete with that peptide for binding to Dxr 13 and not inhibitors with other mechanisms of action. Dxr is easily assayed in a microplate by monitoring oxidation of NADPH, but its substrate, DXP, is very expensive. To overcome this, the possibility of a coupled Dxs-Dxr screen was explored.
Dxs has been shown to be essential in E. coli and very recently in Mtu. 14 It has no human homolog and is a valid target for discovery of new drugs. However, for a very long time, the only inhibitor of Dxs reported was fluoropyruvate, a substrate analogue with weak potency against the enzyme; recently, other inhibitors have been published, but these, too, are very weak (IC50 10–100 µM). 15 Although Mtu Dxs has been well studied, 16 no crystal structure of this protein has been reported. Crystal structures of the E. coli and Deinococcus radiodurans enzymes have been reported, 17 but these do not have inhibitor bound to them. Thus, by our criteria for target evaluation, Dxs, although a valid target, scores lower than Dxr.
Dxs is very difficult to assay because its products are not fluorescent and cannot be monitored spectrophotometrically. Most methods of assaying Dxs are not high throughput. Many use column chromatography to separate product from substrate, 18 often using a radioactive substrate. 19,20 Another avoids chromatography but is cumbersome because it involves heating the product with acid at 80°C to generate a fluorescent product. 21 However, it can be measured indirectly by coupling its activity to Dxr, 22 a method that we used to monitor purification of Dxs.
A coupled Dxs-Dxr assay has the advantage of assaying two targets simultaneously, overcomes the difficulty of assaying Dxs alone, and obviates the need for the expensive Dxr substrate, making an HTS cost-effective. The Dxs-Dxr coupled assay described here is different from many enzyme-coupled assays in which the coupling enzyme is used for signal amplification 23 or to detect a product that is difficult to detect, as in the pyruvate kinase-lactate dehydrogenase coupled systems. 24 In those cases, care is taken not to pick up inhibitors of the coupling enzyme. In a few exceptions (e.g., pathway assays 25 ), these are sensitive to inhibitors of each of the coupling enzymes.
Here, the evaluation of Dxr and Dxs as drug targets is reported. The binding energetics of fosmidomycin, together with pre-existing information on essentiality 3 and structure, 4 give Dxr a high druggability score. A coupled Dxs-Dxr microplate HTS was developed. Despite the fact that Dxr appears to be a more attractive target than Dxs, the intention was to retain the option of picking up Dxs inhibitors, and hence an assay that was equally sensitive to inhibitors of both enzymes was designed. Taking a cue from fosmidomycin binding energetics and the fact that this pocket on the enzyme is druggable, the assay was designed with high NADPH with the aim of finding enthalpically driven inhibitors of Dxr. Because the primary assay cannot distinguish inhibitors of Dxs from those of Dxr, methods to deconvolute the inhibitors were designed. Dxs or Dxr can be individually assayed to test the inhibitors. Alternatively, using a microplate thermofluor assay, one can monitor binding of inhibitors to Dxr or Dxs. Fosmidomycin and fluoropyruvate, inhibitors of Dxr and Dxs, respectively, were used to validate the assays.
Materials and Methods
Fosmidomycin and DXP were purchased from Echelon Biosciences Inc. (Salt Lake City, UT); DXP was also synthesized in house. 26 Ni-NTA resin was from Qiagen (Valencia, CA). All other chemicals were from Sigma-Aldrich Chemical Co. (St. Louis, MO).
Cloning, expression, and purification of Dxs and Dxr
Dxs
M. tuberculosis (Mtu) genomic DNA was used for PCR amplification of the Dxs gene (Rv2682c) using primers designed to introduce a His tag and Nde1 restriction site at the 5′ end and a BamH1 restriction site at the 3′ end. The amplified fragment was subcloned into pET15b vector (Novagen, Madison, WI). The resultant plasmid pAZI0206/tbp10r has the coding region of Dxs plus 20 extra amino acids, MGSSHHHHHHSSGLVPRGSH, at the N-terminus.
For production of Dxs protein, E. coli BL26 (DE3) transformed with pAZI0206/tbp10r was grown in Luria Bertani Broth containing 100 µg/mL ampicillin at 30°C to an A600 ~ 0.6 on a shaker. The culture was further incubated overnight on a shaker at 25°C. Cells were harvested by centrifugation, resuspended in buffer A containing 10 mM imidazole, and lysed using a French press (buffer A: 50 mM phosphate buffer pH 7.9, 500 mM sodium chloride, 1 mM β-mercaptoethanol, 50 µM thiamine pyrophosphate [TPP], and 10% glycerol). The lysate was centrifuged at 100 000 × g for 45 min. The supernatant was loaded onto a Ni-NTA column (Qiagen) pre-equilibrated with buffer A containing 10 mM imidazole. The column was washed with buffer A containing 50 mM imidazole and eluted with a linear gradient of imidazole (50–250 mM) in buffer A in 20 column volumes. The protein concentration of the eluate fractions was monitored, and fractions containing Dxs were dialyzed against 50 mM phosphate buffer pH 7.9, 50 mM TPP, 1 mM 2-mercaptoethanol, and 20% glycerol and stored at –80°C.
Dxr
The Mtu Dxr gene (Rv2870c) was amplified using primers designed to introduce Nc01 and BamH1 restriction sites at the 5′ and 3′ ends, respectively, and a His tag at the 3′ end. The PCR-amplified fragment was subcloned into pET21D (Novagen). The resultant plasmid pAZI0174a coded for the Dxr protein with 13 extra amino acids at the C-terminus: KLAAALEHHHHHH.
E. coli BL21 (DE3) cells transformed with plasmid pAZI0174a were grown in LB containing 100 µg/mL ampicillin at 37°C to an A600 ~0.6. The culture was cooled to 30°C, 1 mM IPTG was added, and culture was allowed to grow overnight at 30°C. Cells were harvested by centrifugation, resuspended in buffer B (buffer B: 50 mM sodium phosphate pH 7.4, 300 mM NaCl), and lysed using a French press. The lysate was centrifuged at 100 000 × g, and the supernatant was loaded onto an Ni-NTA column. The column was washed with 10 column volumes of buffer B with 10 mM imidazole and eluted with a 10–250 mM linear gradient of imidazole in buffer B in a total of 20 column volumes. The final yield was ~1 mg Dxr/1 of cell culture.
Enzyme assays
All assays, except where specified, were performed at 25°C in 100 µL volume in 96-well half-area microplates (Costar 3695; Corning, Corning, NY). NADPH oxidation was monitored by the decrease in A340 using a Spectramax250 (Molecular Devices, Sunnyvale, CA).
Dxs-Dxr coupled assay
The Dxs-Dxr coupled assay reaction contained Dxs (0.42 µg), Dxr (0.047 µg), 0.75 mM glyceraldehyde-3phosphate, 3 mM pyruvate, and 0.225 mM NADPH, 50 mM MOPS-NaOH buffer pH 7.5, 1.5 mM MnCl2, 100 mM NaCl, 0.002% Brij-35, and 2 mM β-mercaptoethanol. This complete reaction is called the positive control.
For testing inhibitors, the two enzymes were preincubated with inhibitors for 15 min in 80 µL of reaction buffer, followed by simultaneous addition of glyceraldehyde-3-phosphate, pyruvate, and NADPH to start the reaction; the background reaction had no pyruvate. For endpoint assays, the final A340 was subtracted from the zero time reading as a measure of signal for both positive control and background. To check if the robustness of the assay was suitable for an HTS, the assay was adapted to 384-well plates: the assay volume was reduced to 60 µL, β-mercaptoethanol was omitted, and 10 mM EDTA was used for the background reaction.
Dxs assays
(a) Dxs coupled assay. For measurement of the specific activity of Dxs or for testing inhibitors of Dxs, its activity was coupled to Dxr; the assay conditions were identical to that described in the “Dxs-Dxr Coupled Assay” section, except that a 10-fold excess of Dxr (0.6 µg) was used. (b) Direct Dxs assay. In cases in which inhibition of Dxs needed to be confirmed or inhibition of Dxs needed to be distinguished from inhibition of Dxr, Dxs was assayed as previously described. 21 Briefly, DXP was estimated by boiling it or the Dxs reaction product in 100 µL volume with an equal volume of 10 mM 3–5 diamino benzoic acid in 5 M phosphoric acid for 15 min. The samples were kept on ice and their fluorescence read (Ex 396 nm, Em 510 nm). A standard curve of DXP was used to estimate the quantity of DXP formed in the assay. The method is not very sensitive, consumes large quantities of DXP (an expensive reagent), and cannot be performed in a microplate.
Dxr assay
The activity of Dxr was monitored spectrophotometrically at 340 nm by following oxidation of NADPH. The reaction was carried out in microplates in 100 µL containing 0.225 mM NADPH, 0.4 mM DXP, 50 mM MOPS-NaOH buffer pH 7.5, 1.5 mM MnCl2, 100 mM NaCl, 0.002% Brij-35, 2 mM β-mercaptoethanol, and Dxr. For testing inhibitors, 0.075 µg of Dxr (specific activity 3 µmoles/min/mg protein) was typically used.
Compound testing
Compounds were dissolved in DMSO, serially diluted in DMSO to a concentration that is 25× the desired test concentration. Four microliters of this stock was used in a 100 µL enzyme assay; compounds were tested in a series of doubling dilution.
ITC
The energetics of binding of fosmidomycin to Mtu Dxr at 25°C was studied using the VP-ITC Microcalorimeter (MicroCal, Northampton, MA). Dxr was dialyzed in a buffer containing 50 mM HEPES-NaOH pH 7.5, 100 mM NaCl, 1.5 mM MnCl2, 2 mM β-mercaptoethanol, and 5% glycerol. Fosmidomycin was dissolved in the dialysate. NADPH (0.25 mM) was added to both protein and fosmidomycin solutions. Titration was carried out by injecting varying volumes (e.g., 20, 10, and 5 µL) of 70 µM fosmidomycin into the cell containing 8.5 µM Dxr. The data were fitted to the single-site–binding model using MicroCal Origin software.
Thermofluor assay
Measurement of shifts in the melting temperature (Tm) of the protein was carried out in 50 mM HEPES pH 7.5, 100 mM NaCl, 1.5 mM MnCl2, and 0.15 mg/mL Dxs (2.3 µM) or Dxr (3 µM). The concentration of ligands used was varied and is indicated in the figure legend. 6.25 × Sypro Orange (Invitrogen, S-6650, Carlsbad, CA) was used as a fluorophore. Assays (50 µL) were set up in 96-well PCR plates (Axygen-PCR-96-FLT-C) and sealed with Microseal B (Biorad, Hercules, CA). The temperature was varied from 25°C to 95°C in steps of 0.5°C, and fluorescence measurements (Ex: 470 nm, Em: 570 nm) were carried out in an iCycler from Biorad. Fluorescence was plotted versus the temperature, In addition, the rate of change of fluorescence with temperature (–dRFU/dT) was plotted versus temperature, and the minimum point of the derivative was taken as the melting point (Tm).
Results
Binding energetics of fosmidomycin to Dxr and druggability
Initial attempts to study binding of fosmidomycin to Mtu Dxr by ITC showed no evidence of binding. However, in the presence of 0.25 mM NADPH, fosmidomycin bound Mtu Dxr with a K D of ~17 nM and with a ΔH of –10.2 ± 0.13 kcal/mol and a ΔS of 1.1 cal mol−1K−1 and a typical binding isotherm was obtained. Three experiments were carried out, and a representative isotherm is shown ( Fig. 2 ). The stoichiometry of binding was 0.6. A stoichiometry of 1 would be expected from the interaction of fosmidomycin with Dxr. 4 The lower value probably reflects inaccuracies in protein estimation or that not all the protein is active and able to bind fosmidomycin. The enthalpic energy of binding is high compared with the entropic contribution, and it appears that the binding is enthalpically driven. Thus, we conclude that Dxr, or at least the fosmidomycin binding pocket in the protein, is druggable and it is likely to pick up novel inhibitors that bind with a high enthalpic component.
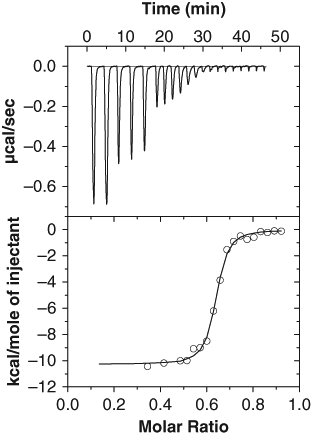
Binding of fosmidomycin to M. tuberculosis Dxr in the presence of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) by isothermal binding calorimetry. Binding was carried out at 25°C at pH 7.5 in the presence of MnCl2 and 0.25 mM NADPH as described. The cell contained 8.5 µM Dxr, and the syringe contained 70 µM fosmidomycin. Two injections of 20 µL were followed by 3 injections of 10 µL and then 15 injections of 5 µL each; the interval between injections was 2.5 min. The data were fitted to the “OneSites model” of the Origin software, giving a stoichiometry of 0.63, K D of ~17 nM, ΔH of –10.2 ± 0.13 kcal/mol, and a ΔS of 1.1 cal mol−1K−1
The absence of a signal when no NADPH was present could either be because fosmidomycin did not bind to Dxr under the conditions used or the heat released on binding was too small to measure. Fosmidomycin has a 10-fold lower K i for E. coli Dxr in the presence of NADPH than in its absence. 12 Also, thermofluor experiments showed a significant change in the melting point (Tm) of Mtu Dxr on addition of fosmidomycin even in the absence of NADPH (later section), indicating that binding does occur.
Because fosmidomycin is a potent inhibitor and even progressed into clinical development as an antibacterial agent, the intention was to bias the HTS to select Dxr inhibitors with a similar mechanism of binding. Hence, it was considered important to have a high concentration of NADPH in the HTS assay.
Enzyme activity of Mtu Dxr and Dxs
Mtu Dxr purified from E. coli had more than 95% purity as judged by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The enzyme oxidized NADPH in the presence of DXP, with highest activity at pH 7.5. Mtu Dxr could use Mg2+, Mn2+, or Co2+ for catalytic activity, as reported earlier 27 ; there is a contrasting report 6 that no Dxr activity was detected in the presence of Mn2+ or Co2+. Enzyme activity varied with concentration of cation, saturating at ~0.5 mM Co2+ or Mn2+ versus ~4 mM Mg2+. The maximum activity obtained with the 3 cations was similar, and Mn2+ was used for most assays.
The specific activity of the protein measured by the Dxr assay was 3 µmoles of NADPH oxidized/min/mg protein. The Km app for DXP with 0.225 mM NADPH and Mn2+ as metal cofactor was ~160 µM, similar to the reported value of 100 µM. 12 The Km app for NADPH was ~10 µM (at 0.75 mM DXP), similar to the reported 3.3 µM. 27 Dxr activity was inhibited by fosmidomycin with an IC50 value of 54 nM.
Mtu Dxs was purified to greater than 90% purity from E. coli cells. Dxs activity was characterized by the fluorescent assay and the Dxs coupled assay; our data were similar to that published in terms of cofactor and cation requirement. 16 The specific activity measured by the Dxs coupled assay was 0.23 µmoles of DXP formed (or NADPH oxidized)/min/mg protein.
Dxs-Dxr Coupled Microplate Assay
Assay design
Three different formats for the Dxs-Dxr coupled assay were tried. In two of these, the assay was performed in two steps: the first step being the Dxs reaction followed by the Dxr reaction in a second step.
In format 1, Dxr, Dxs, and only the Dxs substrates were added initially, in an 80 µL reaction to allow DXP to accumulate. After 60 min at 25°C, the second Dxr substrate, NADPH, was added (20 µL of 1.12 mM), and its oxidation was monitored immediately by measuring A340. In a variation of this, a second format, the Dxs reaction, was stopped by adding fluoropyruvate before adding NADPH to start the Dxr reaction. The rationale behind these two formats was to have a finite and measurable concentration of the Dxr substrate, DXP, at the start of the Dxr reaction and that it may be easier to optimize ratios of Dxs and Dxr. However, no advantage of the two-step reaction format was found, and it was not easier to design the assay to be equally sensitive to Dxr and Dxs inhibitors.
Hence, a one-step format in which all the components were added together was used. This format is logistically much easier, especially for an HTS. The substrates of Dxs, G-3P, and pyruvate and the cofactor TPP were added along with NADPH, substrate of Dxr, at the start of the assay. The other Dxr substrate, DXP, was generated in situ by Dxs during the course of the reaction. This allows the DXP generated by Dxs to be immediately converted to MEP by the Dxr-catalyzed reaction. Under such conditions, it is expected that DXP will not accumulate to high levels, and it should make the assay more sensitive to inhibitors that are competitive with DXP.
In this assay format, different conditions were tested to get the optimum pH, buffer, and salt concentration. A pH between 7.5 and 8, 1 mM TPP, and greater than 1 mM MnCl2 were found to give highest activity ( Fig. 3A – C ). In the optimized buffer and salt conditions (MOPS-NaOH pH 7.5, 1.5 mM MnCl2, 1 mM TPP, 2 mM 2-mercaptoethanol, 0.002% Brij-35, and 0.225 mM NADPH), the Km app of G-3P was 0.14 mM (measured in the presence of 6 mM pyruvate; Fig. 3D ). The Km app for pyruvate was 0.3 mM (measured in the presence of 6 mM G-3P; Fig. 3E ). The intention was to bias the assay to select inhibitors that are either uncompetitive or very potent competitive inhibitors of pyruvate and G-3P, so that they are active even in the presence of high cellular concentrations of the two metabolites. Therefore, in the final assay, G-3P was used at 0.75 mM, 5 times the Km app, and the pyruvate concentration chosen was 3 mM, 10 times the Km app of pyruvate. The Km of NADPH for Dxr is 10 µM. However, NADPH was used at 225 µM to allow selection of inhibitors that, like fosmidomycin, bind better in the presence of NADPH. Also, a high concentration of NADPH was needed to get a reasonable window.
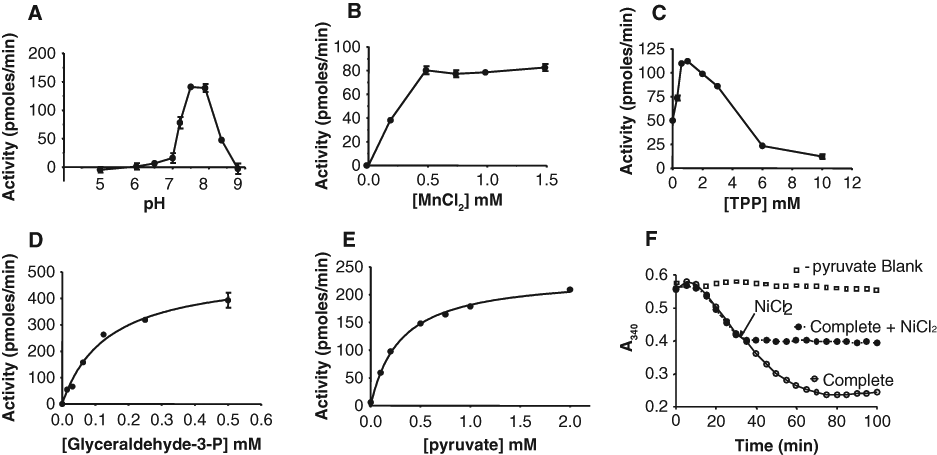
Characterization of the Dxs-Dxr coupled assay. Activity is plotted as a function of (A) varying pH, (B) varying concentrations of MnCl2, (C) varying thiamine pyrophosphate concentration, (D) varying concentration of glyceraldehyde-3-phosphate (in the presence of 6 mM pyruvate), and (E) varying concentration of pyruvate (in the presence of 6 mM glyceraldehyde-3-phosphate). Other assay conditions were as described in the “Materials and Methods” section; values represent the mean ± SD (n = 2). Data in (D) and (E) were fitted to the Michaelis-Menten equation using GraphPad Prism. In (F), a time course of the reaction is shown along with a blank reaction in which pyruvate was left out. At the time indicated, 1 mM nickel chloride was added to the reaction.
A major challenge in the assay development was to optimize Dxs and Dxr concentrations such that the assay was equally sensitive to inhibitors of both the enzymes and dependent on the concentrations of both enzymes. If the two enzyme concentrations are perfectly balanced, with a 50% decrease in either protein concentration, one would expect to see a 50% decrease in enzyme activity. This is easy to achieve with a single enzyme reaction. With a 50% decrease in protein, the activity was not decreased proportionally for either enzyme. As an example, a representative matrix experiment is shown with three concentrations of Dxr (3.5, 7, and 10.5 nM) for each of three concentrations of Dxs (31, 62, and 90 nM; Fig. 4A ). The best achieved was with 62 nM Dxs and 7 nM Dxr. Using this condition as a reference ( Table 1 ), decreasing the concentration of Dxs by 50% resulted in a 42% decrease in activity, and decreasing the Dxr concentration by 50% resulted in a 38% decrease in activity.
Dxs-Dxr Assay: Dependence of Activity on Concentrations of Dxs and Dxr
Dxs-Dxr activity as a function of Dxs concentration was expressed relative to Dxs concentration in row B.
Dxs-Dxr activity as a function of Dxr concentration was expressed relative to Dxr concentration in row E.
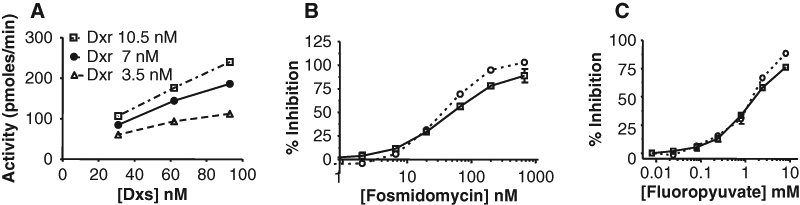
Activity of the Dxs-Dxr coupled assay with varying enzyme ratios (A) and validation of the assay with fosmidomycin (B) and fluoropyruvate (C). (A) Activity as a function of varying concentrations of Dxs at three concentrations of Dxr: 3.5 nM Dxr (Δ), 7 nM Dxr (•), and 10.5 nM Dxr (B). (B) Effect of varying concentrations of fosmidomycin (an inhibitor of Dxr) in the Dxs-Dxr coupled assay versus the Dxr assay (dotted line). The IC50 in the two assays is 79 nM and 54 nM, respectively. (C) Effect of varying concentrations of fluoropyruvate (an inhibitor of Dxs) on the Dxs-Dxr coupled assay or the Dxs coupled assay (dotted line); the IC50 of fluoropyruvate is 2.3 and 1.8 mM, respectively. Values represent the mean ± SD (n = 2 or 3); data were fitted to the 4-parameter logistic.
Terminating the assay
When screening a large number of compounds, it is convenient to stop the enzyme reaction before reading the absorbance, in anticipation of occasional instrument breakdowns. EDTA is typically used to terminate reactions that are dependent on divalent cations. However, in the Dxs-Dxr coupled assay, the addition of EDTA resulted in an extremely rapid oxidation of NADPH, rather than termination of the oxidation, and this could not be explained. It was observed only in Dxs-Dxr reactions using Mn2+ and 2-mercaptoethanol but not if the Mn2+ was replaced by Mg2+. Options considered were to switch to Mg2+ as a cation or find an alternative reagent to terminate the reaction. The latter was chosen; it was found that 1 mM NiCl2 could be used to terminate the reaction ( Fig. 3F ).
Assay validation
The robustness of the assay for an HTS was assessed in a 384-well plate. The average positive control was 0.35 with a coefficient of variation (CV) of 6%; the average background was 0.07, with a CV of 2.6%; and the Z′ 28 was 0.75. The Dxs-Dxr coupled HTS assay was inhibited by both fosmidomycin, a Dxr inhibitor, and fluoropyruvate, a Dxs inhibitor, indicating the assay will detect inhibitors of both enzymes ( Fig. 4B , C ). To compare the sensitivity of the coupled Dxs-Dxr assay versus the individual enzyme assays, the IC50 of the inhibitor in the two assays was compared. The IC50 values of the reference Dxr inhibitor, fosmidomycin, measured in the coupled Dxs-Dxr assay and Dxr assay were 79 and 54 nM, respectively ( Fig. 4B ). The IC50 of the Dxs inhibitor, fluoropyruvate, was 2.3 mM in the coupled Dxs-Dxr assay versus 1.8 mM in the Dxs coupled assay ( Fig. 4C ). The IC50s in the Dxs-Dxr assay are slightly higher than in the assays for the individual enzymes, but the difference is not large, and this degree of variation is of the range of interday variation seen in IC50 determinations. Thus, the Dxs-Dxr assay was considered validated in that it will detect inhibitors of both enzymes with more or less equal sensitivity.
Deconvolution of Inhibitors into Dxs or Dxr Inhibitors
An HTS using this coupled assay will result in selection of inhibitors of both Dxs and Dxr. Dxs inhibitors can be distinguished from the Dxr inhibitors by testing them in assays for the individual enzymes. Alternatively, studying the binding of the compounds to either Dxs or Dxr can be used for deconvolution.
Enzyme assays for deconvolution of inhibitors
A direct Dxr assay was developed and assay conditions are described in the “Materials and Methods” section. For Dxs, either the Dxs coupled assay or the more laborious direct Dxs assay can be used to screen for Dxs inhibitors.
Thermofluor assay for deconvolution of inhibitors
Dxr has a melting temperature (Tm) of 43°C. An increase of 20°C in the Tm of Dxr was observed in presence of 100 mM fosmidomycin ( Fig. 5A ). However, no shift in its melting temperature was observed in the presence of 100 mM fluoropyruvate (data not shown). Similarly, the Tm of Dxs did not shift in the presence of fosmidomycin, but there was a small shift in Tm on addition of 100 mM fluoropyruvate (data not shown). The shift in Dxs Tm was very subtle, and this could be because fluoropyruvate is a weak inhibitor of Dxs. Because compounds that specifically bind to Dxr increase the melting temperature of the Dxr protein, and a similar observation was made with Dxs, this method can be used to distinguish inhibitors of Dxr from Dxs.
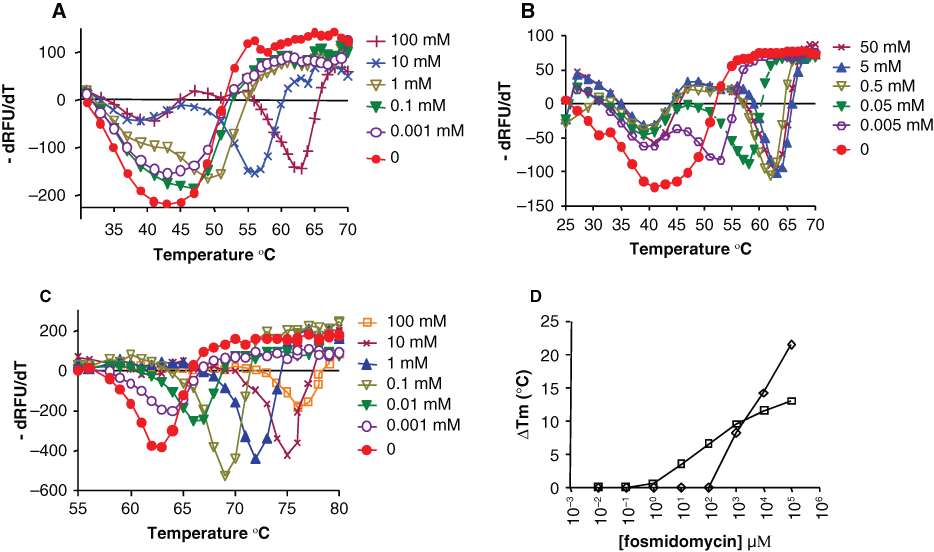
Effect of fosmidomycin or nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) or both on the melting temperature (Tm) of Mtu Dxr. The temperature was varied from 25°C to 95°C in steps of 0.5°C, and fluorescence measurements were carried out in an iCycler instrument (Biorad). 6.25 × Sypro Orange (Invitrogen) was used as a fluorophore. (A) Fosmidomycin was varied from 1 µM to 100 mM. The Tm of Dxr (43°C) increased in the presence of 1 mM fosmidomycin (50.5°C) and kept increasing even up to 100 mM. (B) NADPH was varied from 5 µM to 50 mM. The Tm of Dxr (40.5°C) increased in the presence of 5 µM NADPH and saturated at 62.5°C in the presence of 5 mM NADPH. (C) Mtu Dxr was incubated with 5 mM NADPH and the shift in Tm measured as a function of varying fosmidomycin. The Tm (62.5°C) increased from 10 µM fosmidomycin and kept increasing even up to 100 mM fosmidomycin (Tm of 75.5°C). (D) The increase in Tm on addition of fosmidomycin is plotted versus the concentration of the inhibitor; the Tm in absence of fosmidomycin is used as the comparator. Mtu Dxr (□), Mtu Dxr plus 5 mM NADPH (◊). Note that lower concentrations of fosmidomycin cause a shift in Tm when NADPH is present. The experiment was repeated multiple times with each sample in duplicate, but only one of the replicates from a single experiment is shown; the absolute relative fluorescence units varied between replicates, but the Tm values across replicates were the same.
A concentration response was performed to determine the minimum concentration of fosmidomycin needed to cause a shift in the Tm of Dxr. No shift in Tm was observed from 10 nM to 10 µM fosmidomycin ( Table 2 ). At 100 µM fosmidomycin, there was a subtle change in the temperature profile, but at 1 mM fosmidomycin, there was a distinct increase in Tm to 50.5°C. A further increase in Tm to 56.5°C and 63°C was observed in the presence of 10 and 100 mM fosmidomycin, respectively ( Fig. 5A ; Table 2 ). There appeared to be a fraction of the protein that did not show this increase in Tm, possibly because it did not bind to fosmidomycin.
Mtu Dxr Melting Temperature (Tm) as a Function of Ligand Concentration
The concentration of NADPH used was 5 mM.
It was surprising that no significant shift in Dxr Tm was observed even at 100 µM fosmidomycin, although the compound showed a Kd of 17 nM by ITC and an IC50 of 54 nM in the Dxr assay. With such high affinity, the effect of fosmidomycin on Tm shift should saturate when it is in molar excess over the enzyme concentration (3 µM Dxr in these experiments). Also, although a shift in Tm was observed at very high concentrations (1 mM fosmidomycin), the effect did not saturate even at 100 mM fosmidomycin. A major difference in the Tm shift experiment as compared with ITC and the enzyme assay is that the Tm shifts were carried out in absence of NADPH, whereas both the other assays contained ~0.2 mM NADPH. This suggested that the affinity of fosmidomycin for Dxr increases several fold in the presence of NADPH.
Tm shift experiments were performed to confirm that fosmidomycin has greater affinity to the Dxr-NADPH complex as compared with Dxr alone. The effect of NADPH (5 µM to 50 mM) on melting temperature of DXR was first studied to determine the saturating concentration of NADPH. NADPH caused a concentration-dependent increase in the melting temperature of Dxr ( Fig. 5B ). A significant shift in the Mtu Dxr Tm was observed at 5 µM NADPH, and the shift in Tm saturated at 5 mM NADPH, resulting in a Tm of 62.5°C. The Tm shift of this DXR-NADPH population on addition of fosmidomycin was studied; a proportion of the protein had a Tm of 40.5°C and apparently did not bind NADPH.
The effect of varying fosmidomycin on the Tm of Mtu Dxr was studied in the presence of 5 mM NADPH. A concentration-dependent increase in Tm was observed from 10 µM to 100 mM fosmidomycin, and the effect seems to saturate at 10 mM. At 100 mM fosmidomycin, the Dxr Tm was 75.5°C, a 13°C shift as compared with Dxr-NADPH (Tm 62.5°C; Fig. 5C ; Table 2 ). The lowest concentration at which a shift in Dxr Tm was observed was 10 µM fosmidomycin (4°C shift), whereas in the absence of NADPH, an increase in Tm was observed only at 1 mM fosmidomycin ( Fig. 5D ). This does suggest that the affinity of fosmidomycin for Dxr is greater in the presence of NADPH than in its absence, although it does clearly bind Dxr in the absence of NADPH. Data from the crystal structures support this: a dramatic conformational change in E. coli Dxr is induced by binding of fosmidomycin in the presence of NADPH 29 ; NADPH alone shows a change compared with the apo protein, and this is further enhanced by binding of fosmidomycin. This is not so obvious in the structures of Mtu Dxr. The structural changes may explain the very large Tm shifts observed, which were much larger than we have seen with other proteins.
Discussion
Dxr is an attractive target for the discovery of new antimycobacterial agents. However, assaying Dxr activity in high-throughput format is challenging because of the cost of its substrate DXP. By coupling the activity of Dxr with Dxs, also a valid drug target, the expense of directly assaying Dxr can be circumvented and two targets screened simultaneously in a cost-effective manner.
A major challenge was selecting enzyme concentrations of Dxs and Dxr to make the assay sensitive to inhibitors of both enzymes. Because of the substrate concentrations used, the assay is biased to select for uncompetitive or very potent competitive inhibitors of pyruvate or G-3P, uncompetitive or very potent competitive inhibitors of NADPH, and inhibitors competitive with DXP. Most of the assay optimization was performed with a background reaction in which one of the Dxs substrates (pyruvate) was left out of the reaction. The potential for an HTS was estimated by modifying the assay conditions and using EDTA in the absence of 2-mercaptoethanol for the background reaction. We have also shown that NiCl2 can be used to terminate the reaction, allowing two options of background reaction for an HTS.
Inhibitors of the coupled assay can be deconvoluted into Dxr or Dxs inhibitors by screening compounds in the individual enzyme assays, measuring the Tm shift in a thermofluor assay, or binding to either protein by ITC or other biophysical methods.
Interpretation of the Dxs coupled assay can be difficult because very potent Dxr inhibitors (e.g., nM IC50s) would inhibit it at high concentrations. In such cases, the direct Dxs assay could be used to confirm inhibition of Dxs. Also, when inhibitors are promiscuous or nonspecific, 30 they may appear to inhibit both Dxs and Dxr. In such cases, confirmation of binding in the thermofluor assay is invaluable and could be used as an orthogonal confirmation of inhibition.
Binding of a ligand (e.g., a substrate or inhibitor) to a protein usually stabilizes protein structure and results in an increase in the melting point of the protein. Thus, a shift in the Tm of a protein in the presence of a ligand is a good indication of binding. This assay can be performed in a microplate in quite high throughput. An interesting question is what concentration of inhibitor should be used to study the binding of a novel compound. The shift in melting temperature with ligand is dependent on both the concentration of protein and its affinity for the ligand. With 3 µM protein, inhibitors can be tested at standard concentrations of 10 µM and 50 µM or 10× the IC50 of the inhibitor for less potent inhibitors. In addition, learning from the experience of fosmidomycin, it would be advisable to test the binding of compounds to Dxr in the presence and absence of NADPH. The degree of shift in Tm varies with ligand and is influenced by the entropic component of the binding, as well as by the concentration of the ligand relative to its affinity. Hence, it is not always possible to see a shift in Tm on binding of a ligand. But when shifts are observed, it is an indication of binding and can be used as a confirmatory assay to avoid false hits and artifacts that may occur in enzyme assays. Promiscuous inhibitors that denature the enzyme show atypical or no thermal transitions. These Tm shift assays can also be designed to indicate the mechanism of binding of the compound, for example, if the inhibitor binds to Dxr alone or a Dxr-NADPH complex.
The Dxs-Dxr coupled microplate assay described here has an advantage over a previously described HTS assay for Dxr 13 in that it should detect a wider range of inhibitors because it measures enzyme activity and not competition for binding of a peptide. It is a homogenous assay, requires very few manipulations, and is HTS compatible. It is equally sensitive to inhibitors of Mtu Dxs and Dxr. The principle of a coupled assay is applicable to other enzymes in this pathway and also to unrelated enzyme targets. This assay can be used to find novel starting points for discovery of new antituberculosis agents.
Footnotes
Acknowledgements
We acknowledge Sudha Ravishankar and Sunita Ramesh for cloning Mtu Dxs and Dxr, respectively, and Vidya Prasanna Kumar for purification of Dxs. DXP was synthesized in house by Sanjeev Kulkarni, P. Lakshmipathi, and Manoj Kale. We acknowledge Dwarakanath Prahlad and Gayathri Srinivasa for determination of Z′ and B. R. Srinivasa for scientific discussions during the course of this work.