
Abstract
The protective antigen (PA) of Bacillus anthracis is a secreted protein that functions as a critical virulence factor. Protective antigen has been selected as a biomarker in detecting bacterial infection. The in vitro selection method, systematic evolution of ligands by exponential enrichment (SELEX), was used to find single-stranded DNAs that were tightly bound to PA. After 8 rounds of the SELEX process with PA, 4 different oligonucleotides (referred to as aptamers) that contain a 30-residue ssDNA sequence were identified. Dissociation constant (Kd) values with Cy3-attached aptamers were determined via fluorophotometry to be within a nanomolar range. The authors attempted to visualize the detection of PA using an aptamer-based enzyme-linked immunosorbent assay method, which has proven to be successful within a nanomolar Kd value range. Furthermore, 2 of the 4 aptamers exhibited specificity to PA against bovine serum albumin and bovine serum. The results of this study demonstrate the analytical potential of an oligonucleotide-based biosensor for a wide variety of applications, particularly in diagnosing disease through specific protein biomarkers.
Introduction
T
The rapid and sensitive detection of anthrax is of paramount importance not only for human health but also for the establishment of homeland security against bioterrorism. The secreted proteins (PA, LF, and EF) as well as a spore form of B. anthracis can serve as a potential biomarker. Current detection of anthrax includes the use of europium nanoparticles 3 and of square wave voltammetry 4 ; however, the most common detection method of protein targets occurs through an antibody-based enzyme-linked immunosorbent assay (ELISA). 5 The problem with this method is that the antibodies used exhibit sensitivity to temperature variation, undergo irreversible denaturation, and have limited lifetimes.
Systematic evolution of ligands by exponential enrichment (SELEX) is a technology using a random pool of materials to select what tightly bind to or to inhibit target molecules such as proteins, peptides, dyes, amino acids, and drugs. 6,7 The SELEX process can provide a rapid isolation of high binding oligonucleotides to the targets from random oligonucleotides. The binding oligonucleotides are referred to as aptamers, which have several advantages over antibodies. For example, the aptamers are easy to synthesize, simple to chemically modify, are minimally immunogenic and small in size, and do not require animals for synthesis. Because of these advantages, the development of aptamers as therapeutic, diagnostic, and analytical reagents becomes significant. 6,7 Consequently, aptamer-based analytical methods provide a new alternative way for the detection of virulent toxins.
In this study, we used the SELEX technique to find single-stranded DNA (ssDNA) aptamers for PA63, which will result in the detection of anthrax. Given PA63 is the key protein for assisting the internalization of LF and EF, the detection of PA63 would be of central importance to discovering early toxin release. Binding affinity and specificity of the aptamers were quantified by dissociation constant (Kd) measurements. An ELISA method was established to visualize binding to PA63. In addition, computational analysis was performed to predict secondary structures that may contribute to the binding site for PA63.
Materials and Methods
Construction of plasmid for PA63 protein
The plasmid pET28a-PA63 was constructed by polymerase chain reaction (PCR) using previously constructed pET22b-PA83 8 as a template. The PA63 was cloned into pET28a at BamHI and HindIII sites, and the cloning was confirmed by DNA sequencing.
Expression and purification of PA63
The expression plasmid was transformed into Escherichia coli BL21(DE3) using the heat shock method. Transformed bacteria were grown at 37°C in Luria-Bertani (LB) medium containing 100 µg/mL kanamycin until the optical density (OD) at 600 nm of the culture reached 0.7. Expression of PA63 was induced by the addition of 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) and maintained overnight at 18°C. Since PA63 is insoluble, purification was done under dena-turing conditions with on-column refolding as reported previously. 8 The purity of PA63 was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were visualized with Coomassie blue R-250 strain and sizes were estimated with protein standards. Approximately 1.0 mg of PA63 was obtained from 1 L of LB medium culture.
Preparation of single-strand DNA library
A 60-base single-stranded DNA library containing 30 bases of randomized sequence (N)30 5′dGCGCGGATCCCGCGC(N30)CGCGCGAAGCTTGCG3′ was obtained from IDT, Inc. (Coralville, IA). This template DNA library was amplified by PCR with the corresponding primers: a forward primer 5′Biotin-GCGCGGATCCCGCGC 3′ with BamHI site and a reverse primer 5′dCGCAAGCTTCGCGCG 3′ with HindIII site. To obtain ssDNA, we used the previously established method. 9 A 6 M urea polyacrylamide gel was used to separate the biotinylated ssDNA from the amplified DNA, which was incubated with streptavidin (purchased from NEB, Inc., Ipswich, MA) in a way that binds to the biotinylated ssDNA. The complex of the biotinylated ssDNA and streptavidin migrates much slower in the urea gel than the other strand of ssDNA that does not contain biotin. The lower DNA band that does not contain the complex of biotin and streptavidin was excised from the gel to obtain ssDNA using a crush-and-soak method, 10 followed by ethanol precipitation. The lower DNA band, corresponding to the antisense strand generated by reverse primer, was chosen to avoid any possibility that the biotin attached to the other strand could affect the binding of the oligonucleotide to PA63.
In vitro selection
The selection of PA63 binding ssDNA aptamers was obtained using a membrane filtration method with ultrafiltration (a 100-kDa cutting membrane containing Microcon; Millipore, Billerica, MA) and a Micro high-speed refrigerated centrifuge (Micro 17, Hani Science, Seoul, Korea). PA63 was incubated with 10 mM Tris-HCl (pH 8.0) for 30 min at room temperature with varied ssDNA concentrations and at various time periods ( Table 1 ). Centrifugation was carried out to remove unbound DNA through ultrafiltration (12,000 rpm for approximately 1 min). The DNA participating in complex formation were amplified by PCR with nonbiotinylated primers and loaded on a 2.5% agarose gel to confirm the presence of DNA and to remove any side products that may be generated by PCR. The extracted DNA was amplified with biotin-attached forward primer to separate ssDNA on the urea gel as described above. This process was repeated 8 times with varying conditions aimed at increasing the specificity of the ssDNA for the target protein ( Table 1 ).
PA63-ssDNA Binding Conditions for 8 Selection Rounds
Cloning and sequencing
After 8 rounds of the SELEX process, the final ssDNA was amplified by PCR, and products were purified using the crush-and-soak method. Ligation was carried out with the plasmid pET28a (Novagen, Madison, WI) and the ssDNA, both of which were treated with BamHI and HindIII. After E. coli DH5alpha transformation with the ligated plasmids, several colonies were picked and sequenced using a T7 terminator primer.
Determination of dissociation constants (Kd)
To obtain fluorescently labeled aptamers, PCR was carried out using a biotinylated forward primer and Cy3-attached reverse primer. A 6 M urea–polyacrylamide gel was used to separate Cy3-attached ssDNA from the other biotinylated ssDNA that forms a complex with straptavidin after a 30-min incubation period. The Cy3-attached ssDNA was obtained from the gel using a crush-and-soak method. A fixed concentration (0.5 µg/well) of 90 µL PA63 protein was incubated with 20 mM HEPES (pH 7.4) for 30 min at room temperature and was loaded onto a 96-well black immunoplate (SPL, Kyounggi-do, South Korea) and incubated for 1 h with shaking to immobilize PA63 on the plate. Wells containing PA63 were washed with 90 µL Tris-buffered saline Tween-20 (TBST), containing 0.5 M Tris base, 9% NaCl, and 0.5% Tween-20 (pH 8.4), 3 times and incubated with 90 µL of 2% bovine serum albumin (BSA) for 1 h. TBST was again used to wash the wells 5 times. The ssDNA (90 µL) was added to wells in the 96-well plate at various concentrations and incubated for 1 h with shaking. Excess ssDNA was removed by washing with 90 µL of 20 mM HEPES (pH 7.4) 3 times. Finally, the ssDNA:PA63 complex was placed in 90 µL of 20 mM HEPES (pH 7.4). The fluorescent intensities of the Cy3-attached ssDNAs were measured by a Molecular Devices (Sunnyvale, CA) Gemini EM fluorescence microplate reader (excitation: 545 nm; emission: 572 nm).
Determination of dissociation constants (Kd) using ELISA method
The dissociation constants (Kd) were also determined using an ELISA method for PA63. A biotin-labeled aptamer and streptavidin horseradish peroxidase (HRP) antibody were used for this method. To obtain the biotin-labeled aptamer, asymmetric PCR was carried out with a forward primer (10 µM) and a biotinylated reverse primer (100 µM). Six percent native-PAGE was used to separate biotin-bound ssDNA from the unbound ssDNA according to different migrations on the gel. The biotin-attached ssDNA was obtained from the gel using a crush-and-soak method. PA63 protein was incubated with 20 mM HEPES (pH 7.4) for 30 min at room temperature to allow oligorimerization formation. A fixed concentration (0.5 µg/well) of 90 µL PA63 in 20 mM HEPES (pH 7.4) was loaded onto a 96-well polystyrene plate (SPL) and incubated for 1 h with shaking. Wells containing PA63 were washed with 90 µL TBST 3 times and incubated with 90 µL of 5% skim milk for 1 h. The wells were then washed 5 times with TBST. Biotinylated ssDNA (90 µL) was added to the wells at various concentrations and incubated for 1 h with shaking. Excess ssDNA was removed by washing with 90 µL TBST 3 times. Then, 90 µL streptavidin HRP (1:1000 in TBST; BD Biosciences, Franklin Lakes, NJ) was added to the individual wells and incubated for 1 h with shaking. After washing with 90 µL TBST 3 times, a color-developing reaction with 90 µL TMB substrate solution (3,3′, 5,5′-tetramethyl benzidine; R&D Systems, Minneapolis, MN) was carried out. After 15 min, the reaction was terminated with the addition of 90 µL of 1M H2SO4, and the optical densities were measured with the Biotrak multiwell plate reader (Amersham Biosciences, Piscataway, NJ) at 450 nm.
Determination of specificity
To determine the specificity, selected Cy3-attached ssDNA aptamers were obtained by PCR and urea-PAGE. Selected aptamers were tested for their ability to bind BSA and bovine serum (Invitrogen, Carlsbad, CA), and binding specificity was tested with BSA and bovine serum versus PA63. The specificity assay was conducted using a 96-well black immunoplate (SPL). Then, 1 µg of 90 µL proteins in 20 mM HEPES (pH 7.4) was loaded into each cell on the black immunoplate and incubated for 1 h with shaking. Wells containing protein were washed with 90 µL TBST 3 times and incubated with 90 µL of 5% skim milk for 1 h. TBST was again used to wash the wells 5 times. Then, 90 µL of 20 mM HEPES (pH 7.4) ssDNA was added to the wells at fixed concentrations (100 nM) and incubated for 1 h with shaking. Excess ssDNA was removed by washing with 90 µL of 20 mM HEPES (pH 7.4) 3 times (note that we used HEPES buffer in place of TBST). Finally, 90 µL of 20 mM HEPES (pH 7.4) was added to the wells containing the ssDNA:PA63 complex. The fluorescent intensities of the Cy3-attached ssDNAs were measured with a Molecular Devices Gemini EM fluorescence microplate reader (excitation: 545 nm; emission: 572 nm).
Prediction of secondary structure
Secondary structure analysis of candidate aptamers was performed by free-energy minimization using the M-fold program. 11
Results and Discussion
Anthrax PA63 protein was purified as a target to obtain aptamers from a random ssDNA library using in vitro SELEX. The starting library was composed of a 60-bp-long oligonucleotide ssDNA containing a 30-bp randomized oligonucleotide insert flanked on either side by different 15-bp primer sequences. To increase binding affinity and specificity, NaCl was added to the incubation solution after the fifth round ( Table 1 ) of the in vitro selection. After the eighth round of the in vitro selection, we picked up 15 colonies and had all of them sequenced. We found a total of 10 successful sequences and acquired 4 independent inserts; we call these 4 aptamers BH-1, -2, -3, and -4 ( Table 2 ). It should be noted that the same sequence contents were observed in different colonies. For example, in the aptamer BH-3, we found the BH-3 nucleotide sequence 7 times, whereas other aptamer sequences appeared only once.
Summary of Aptamer Sequences, Frequencies, and Kd Values
ELISA, enzyme-linked immunosorbent assay.
Fifteen sequencing events were attempted, and 10 total successful sequences were acquired.
Frequency represents the number of redundant sequencing.
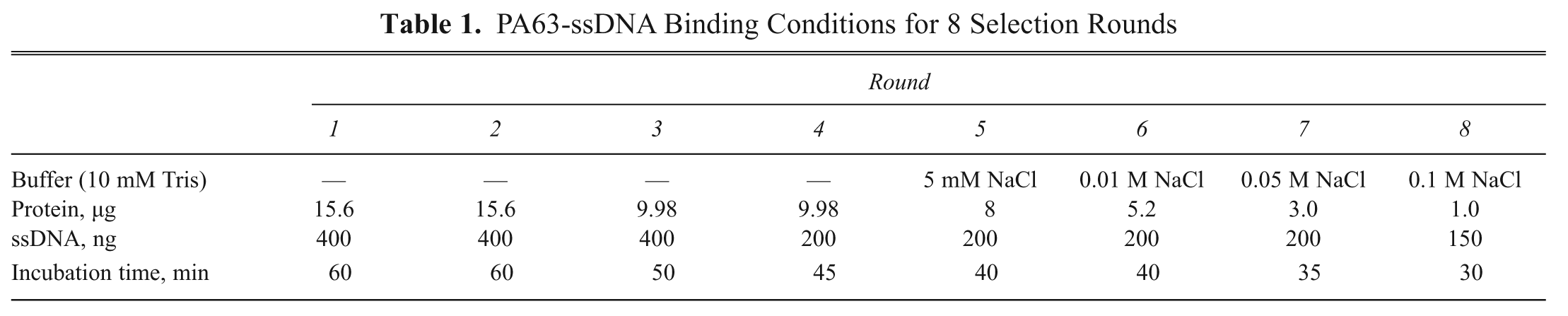
The Kd values of the 4 different aptamers were determined using fluorophotometry. Binding reactions were performed with a constant concentration of PA63 as the target in a 96-well black plate and varied concentrations of each Cy3-attached aptamer. After several washing steps (see Materials and Methods), the PA63 protein-bound aptamers were measured with fluorescent readings and plotted against the aptamer concentration used in the binding reaction ( Fig. 1 ). The Kd values were obtained from nonlinear fitting of the saturation binding curve. The determined Kd values of the 4 aptamers were all within the nanomolar range ( Table 2 ). These results suggest that all aptamers are good candidates for detecting PA. In particular, the Kd values of BH-2, -3, and -4 are in the low nanomolar range, whereas BH-1 has a higher Kd value (38.17 nM). These low Kd values suggest that oligonucleotides can be an effective detector of PA. Furthermore, SELEX technology appears to be a reliable tool for generating molecules that detect macromolecules. In a previous study to find a detecting material using SELEX, Tang et al. 12 determined that ssDNA aptamers bind to a target protein, abrin toxin, with Kd values between 28 and 130 nM.
Determination of binding affinities. The binding affinity of each aptamer was estimated by measuring the binding constant as described in Materials and Methods. Binding reactions were performed with a constant concentration of PA63 heptameric protein in a 96-well black plate as the target and varied concentrations of each Cy3-attached aptamer (0-1000 nM). After several washing steps, the PA63 protein/bound aptamers were measured with fluorescent readings and plotted against the aptamer concentration used in the binding reaction. These data are representative of 1 of 3 independent experiments, and the binding affinity, which was measured as Kd, was obtained from nonlinear fitting of the saturation binding curve.
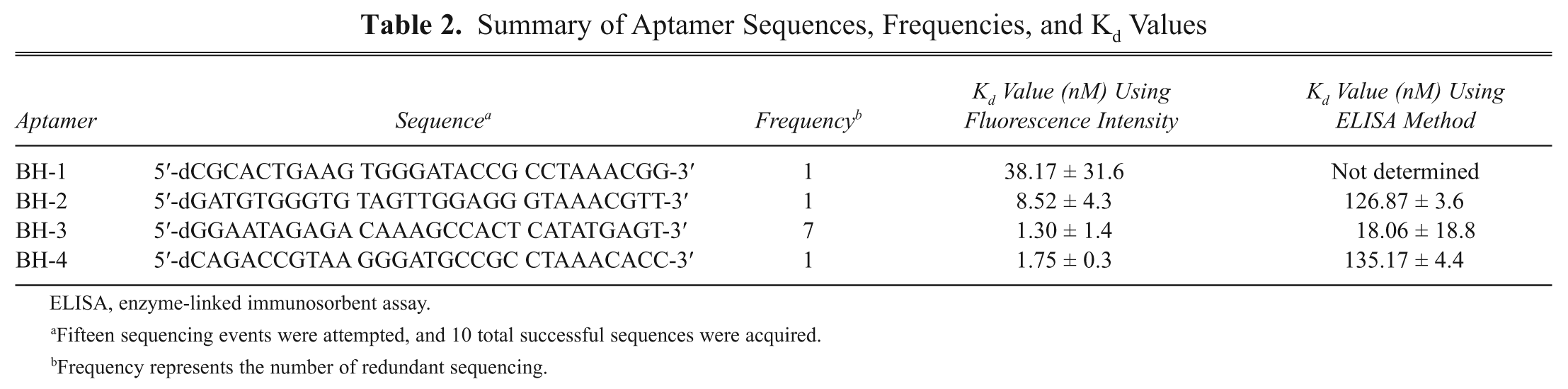
We developed an ELISA method to visualize the binding between PA and aptamers. In this method, PA63 was immobilized on a plate, and then the individual aptamer was added in a concentration-dependent manner (see Materials and Methods). Color development using the TMB reaction coupled with HRP-conjugated proteins containing streptavidin proved to be successful. In this method, the aptamers were used to detect PA in place of antibodies; antibodies are the typical molecule used for detecting targets in the ELISA method ( Fig. 2A ). Figure 2B shows the results of the aptamer-based ELISA method. The data clearly reveal that as the aptamer concentration increased, the intensity of the color increased. We also quantified color intensity to determine Kd values. The quantitative values fit well into a single-substrate binding equation, which was used to determine the Kd value by a direct interaction between an aptamer and PA63. However, the Kd values obtained by ELISA were higher than the values obtained by the Cy3 fluorophore detection method. The high Kd values obtained through the ELISA method can be explained by the fact that the ELISA method needs more binding steps to visualize the detection; for example, the ELISA method requires the HRP-conjugated streptavidin step, but this step is not necessary in the Cy3 fluorophore detection method. We previously employed a similar ELISA method to detect proteins using oligopeptides and obtained reliable binding affinity results, 13 supporting the proposal that this ELISA method is useful in visualizing aptamers binding to a target protein.
Aptamer-based enzyme-linked immunosorbent assay (ELISA). (
The determination of aptamer specificity for PA63 is of keen interest because these aptamers can be used as detecting materials. We attempted to determine aptamer specificity using the aptamer-based ELISA method by comparing the binding of the aptamer to PA63, BSA, and bovine serum. In particular, we used bovine serum because the serum more closely mimics physiological conditions. Prior to measuring the binding with BSA and bovine serum, as control experiments, 5% skim milk was used in the ELISA method to test whether the skim milk binds to the found aptamers; unlabeled aptamers were also used to check whether any signals could be detected. It was found that no noticeable detection against 5% skim milk and unlabeled aptamers was observed. Next, we tested the binding specificity with BSA and bovine serum. Although we were unable to obtain binding specificity for BH-1 and BH-3 because both BH-1 and BH-3 were able to bind BSA and bovine serum just as well as PA63, BH-2 and BH-4 displayed exquisite binding specificity against BSA and exhibited reasonably good binding specificity against bovine serum, as shown in Figure 3 . These results suggest that we may use the aptamers BH-2 and BH-4 as detecting materials for PA.
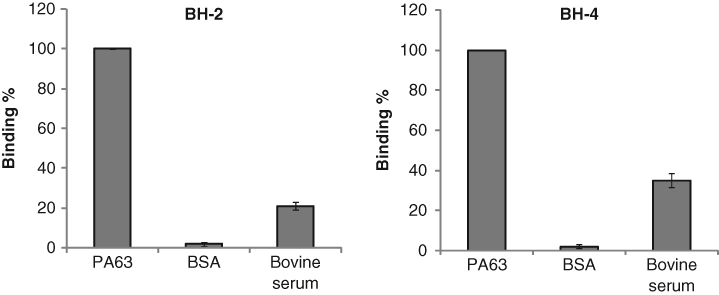
Determination of binding specificity of the aptamers against bovine serum albumin (BSA) and bovine serum using the enzyme-linked immunosorbent assay method. Two hundred nM of each ssDNA aptamer was incubated with a constant concentration of proteins (PA63, BSA, and bovine serum).
To further explore the 2 aptamers that exhibited a low nanomolar range binding affinity with specificity, we hypothesized that the binding sites of the aptamers would be predicted by in silico analysis if the computer program could predict secondary structures. To investigate this possibility, the computer program MFold was used to predict secondary structures. 11 Although we were unable to acquire a secondary structure of BH-2 with a negative free energy during in silico analysis, the program was able to display a stem-loop secondary structure of BH-4 with a free energy of −2.34 kcal/mol, where the sequence containing the 8-residue ssDNA (8-mer) was 5′-d(CCGTAAGG)-3′ ( Fig. 4 ). We determined the Kd value of the 8-mer with a predicted free energy of −0.69 kcal/mol. Measuring the fluorescent intensity of the Cy3-attached 8-mer, the Kd value was determined to be 1.99 ± 1.5 nM, which lines up with values obtained for BH-2. With this observation, the 8-mer appeared to be responsible for binding sites. Yet, when we attempted to determine the binding specificity, the 8-mer could bind to BSA just as well as PA63. In addition, the 8-mer binding to PA63 was only 2 times better than to proteins in bovine serum. These results indicate that the 8-mer is not specific in some way, unlike the 30-residue ssDNA sequence (30-mer) of the aptamers. This observation suggests that a large portion of the 30-mer may contribute to PA63 binding or that the secondary structure prediction is insufficient in identifying the binding sites of the nucleotide aptamer. For example, Schultze et al. 14 demonstrated that the binding of an aptamer for the protein, thrombin, was explained by the 3-dimensional structure of the aptamer. Further study is necessary to improve our understanding of the binding sites for our found aptamers.
Predicted secondary structure for the BH-4 aptamer.
In conclusion, we demonstrated the production of aptamers that specifically bind to PA63 using SELEX technology. The measurement of Kd values by fluorophotometry revealed that aptamers tightly binding to PA have Kd values within a low nanomolar range. In addition, we demonstrated that detection of the target protein can be visualized using the aptamer-based ELISA method, which can be added to a repertoire of protein detection methods. The observation that two aptamers can bind to PA specifically against bovine serum within similar physiological conditions reflects the possibility for the application of aptamers in bacterial B. anthracis detection. In addition, this aptamer-based detection method can be globalized as an oligonucleotide-based biosensor of specific protein biomarkers. Furthermore, discovery of aptamers against PA63 could contribute to overcoming the problems present in current detection methods, such as antibody instability, and thus may advance public health and homeland security against anthrax threat. In a future investigation, we want to enhance our knowledge of the interaction between aptamers and target proteins and increase the binding sensitivity using the resulting aptamers. These experiments are currently under way in our laboratories.
Footnotes
Acknowledgements
This work was supported by the research fund of Hanyang University (HYU-2008-5); a grant (#20080401034006) from the BioGreen 21 program of the Rural Development Administration, the Republic of Korea (MYY); and by funds from the Baylor University Young Investigator Development Program Award (SKK).