
Abstract
The flaviviral RNA-dependent RNA polymerase (RdRp) is an attractive drug target. To discover new inhibitors of dengue virus RdRp, the authors have developed a fluorescence-based alkaline phosphatase–coupled polymerase assay (FAPA) for high-throughput screening (HTS). A modified nucleotide analogue (2′-[2-benzothiazoyl]-6′-hydroxybenzothiazole) conjugated adenosine triphosphate (BBT-ATP) and 3′UTR-U30 RNA were used as substrates. After the polymerase reaction, treatment with alkaline phosphatase liberates the BBT fluorophore from the polymerase reaction by-product, BBTPPi, which can be detected at excitation and emission wavelengths of 422 and 566 nm, respectively. The assay was evaluated by examining the time dependency, assay reagent effects, reaction kinetics, and signal stability and was validated with 3′dATP and an adenosine-nucleotide triphosphate inhibitor, giving IC50 values of 0.13 µM and 0.01 µM, respectively. A pilot screen of a diverse compound library of 40,572 compounds at 20 µM demonstrated good performance with an average Z factor of 0.81. The versatility and robustness of FAPA were evaluated with another substrate system, BBT-GTP paired with 3′UTR-C30 RNA. The FAPA method presented here can be readily adapted for other nucleotide-dependent enzymes that generate PPi.
Introduction
D
The flavivirus NS5 protein contains both methyltransferase and RNA-dependent RNA polymerase activities that are essential for viral propagation. 8-10 The N-terminal 1-272 amino acids form the S-adenosyl-L-methionine-dependent methyltransferase domain, which methylates the RNA cap to form N7-methyl-guanosine and 2′-O-methyl adenosine, resulting in a structure similar to the mammalian mRNA cap. 10,11 The C-terminal part of NS5 (residues 273-900) encodes the RNA-dependent RNA polymerase responsible for synthesizing RNA copies of both plus and minus polarity. 12 The DENV-3 NS5 RNA-dependent RNA polymerase (RdRp) structures, both in apo and nucleotide-complexed forms, have been determined, 13 enabling rational drug design and potentially aiding inhibitor development. The absence of RdRp activity in host cells makes it an attractive target for antiviral drug development.
Several reports have described the identification of flavivirus NS5 inhibitors using high-throughput screening (HTS) as the starting point. The assays are usually based on radioactivity (e.g., scintillation proximity assays in solid phase). 4,14 We have previously developed a DENV RdRp scintillation proximity assay for HTS using [3H]GTP and poly(C)/oligo(G)20 as the substrate and RNA template, respectively, resulting in the identification of an N-sulfonylanthranilic acid inhibitor with an IC50 at 0.7 µM. 4 However, the drawbacks of radioactive assays are requirements for safety precautions and potential harm to both the scientists and environment. To identify novel inhibitors of DENV RdRp with a different readout, we report here the development of a fluorescence-based polymerase assay (FAPA) with improved robustness. The method is similar to the polymerase assay reported by Bhat et al. 15 and Kozlov et al. 16 To achieve a robust assay suitable for HTS, several parameters, including optimal assay buffer components, enzyme/substrate ratios, reaction times, signal stability, and reaction kinetics, were investigated. The signal-to-noise (S/N) ratio and Z factor were used to evaluate the robustness and reproducibility of the assay. The assay was validated by IC50 determination using a specific adenosine nucleotide triphosphate analogue and 3′dATP as inhibitors. A pilot screen consisting of 40,572 compounds in 384-well plates was used to determine the performance of the assay. Our FAPA has proved to be feasible and suitable for HTS for inhibitor finding, with a Z factor of 0.8. In addition to assay development for compound screening, we also evaluated FAPA with an additional nucleotide-RNA substrate system comprising BBT-GTP and 3′UTR-C30. The versatility of FAPA was demonstrated by evaluating it with other NS5 RdRp enzymes from DENV-2 and 3. Our results indicate that FAPA is widely adaptable for the development of assays for nucleotide-dependent enzymes that generate PPi, such as polymerases and transferases.
Materials and Methods
Common assay reagents and enzymes used in molecular cloning were purchased from Sigma-Aldrich (St. Louis, MO), Fisher Scientific (Waltham, MA), Roche Diagnostics (Indianapolis, IN), Stratagene (La Jolla, CA), Invitrogen (Carlsbad, CA), and New England Biolabs (Ipswich, MA). AttoPhos® kit was purchased from Promega (Madison, WI). QIAamp viral RNA mini kit, Qiagen plasmid maxi kit, QIAguick gel extraction kit, and QIAquick PCR purification kit were purchased from Qiagen (Valencia, CA). Superscript III reverse transcription kit and Zero Blunt TOPO PCR cloning kits were from Invitrogen. BBT-ATP and BBT-GTP were synthesized by and purchased from Jena Bioscience GmbH (Jena, Germany). The 3′dATP and 3′dGTP were purchased from Trilink Biotech (San Diego, CA), and RNA substrates 3′UTR-U30 (5′-bio-U30-AACAGGUUCUAGAACCUGUU-3′) and 3′UTR-C30 (5′-bio-C30-AACAGGUUCUAGAACCUGUU-3′) were purchased from Dharmacon (Lafayette, CO). These hairpin self-priming RNAs contain 30 nucleotide-long incorporation regions fused with the 3′UTR sequence (negative strand in italics, complementary positive strand underlined). The dNTP mix was purchased from Roche Diagnostics, and 96- and 384-well black plates were from Corning Costar (Corning, NY). BBT fluorescence was monitored by either Tecan SafireII or Infinite® M1000 plate reader (Tecan, Durham, NC). The compound library used in this study contained compounds that were selected for structural diversity and for adhering to Lipinski’s rule-of-5 and were obtained from various companies.
DENV NS5 cloning and expression
Total RNA was extracted from tissue culture media supernatant containing D4MY01-22713 virus using the QIAamp Viral RNA kit following the manufacturer’s instructions. The purified RNA was reverse-transcribed with Superscript III using the primer 5′-CAATGGTCTCTTTGGTGTTTG-3′. After reverse transcription, the primers 5′-CATATGGCTAGCGGAACTGGGACCACAGGA-3′ and 5′-GCGGCCGCTTACAGAACTCCTTCACTCTC-3′ were used to amplify D4MY01-22713 NS5. The amplified PCR band was subsequently cloned into a zero blunt TOPO PCR cloning vector. After verification of the DNA sequence, the TOPO vector was cut with NheI and NotI restriction enzymes to release the DENV-4 NS5 fragment and cloned into pET28a cut with the same enzymes. DENV-2 NS5 (NGC strain) and DENV-3 (D3MY00-22366 strain) were cloned with similar procedures. The ligation reaction was transformed into Top10 cells (Invitrogen). Protein expression and purification were performed as previously described. 13 The recombinant protein was stored in 20 mM Tris-HCl (pH 7.0), 500 mM NaCl, 10% glycerol, and 10 mM 2-mercaptoethanol at −80 °C until use.
Optimization of alkaline phosphatase reaction on AttoPhos® polymerase by-product mimic
The phosphatase hydrolysis reaction by calf intestinal alkaline phosphatase (CIP) on AttoPhos® (2′-[2-benzothiazoyl]-6′-hydroxybenzothiazole phosphate [BBTPi]; Promega) 17 was optimized in a 10-µL reaction containing 0.1 to 0.2 µM AttoPhos®, 0.1 to 10 nM CIP in 10 to 500 mM deoxyethanolamine (DEA), 200 mM NaCl, and 25 mM MgCl2. The reaction was incubated at room temperature, and the release of BBT was continuously monitored at ex/em 422/566 nm for up to 16 h.
BBT-ATP utilization by DENV-4 NS5 RdRp, time course, buffer optimization, and kinetics
The RNA template, 3′UTR-U30 or 3′UTR-C30, was resuspended to 200 µM in a buffer consisting of 50 mM Tris-Cl (pH 8.0) and 150 mM NaCl in 0.1% diethyl pyrocarbonate (DEPC) water. The solution was incubated at 55 °C to 60 °C for 5 min and placed at room temperature to allow the formation of the intramolecular hairpin. Polymerase activity was investigated with 50 nM NS5 in a time course experiment in 50 mM Tris-HCl (pH 7.0), 2 mM dithiothreitol (DTT), 10 mM KCl, and 1 mM MnCl2 in a 30-µL reaction in a 96-well, half-well plate with either 20 or 100 nM RNA and 2 µM BBT-ATP. At intervals ranging from 0 to 300 min, 20 µL of stop buffer (200 mM NaCl, 25 mM MgCl2, 1.5 M DEA) containing 25 nM CIP was added to terminate the NS5 polymerase reaction and to allow hydrolysis of BBTPPi by CIP. The released BBT was monitored after a 1-h incubation.
The optimization of the NS5 and RNA concentrations was carried out in 30-µL reactions in 96-well, half-well plates. Reactions containing 2.5 to 50 nM NS5 were tested with 10, 20, or 50 nM of RNA at a constant BBT-ATP concentration of 2 µM. The buffer composition was optimized by investigating the effect of 0 to 300 mM NaCl or KCl, 0 to 16 mM MgCl2, 0% to 0.03% (w/v) Triton X-100, 0 to 5 mM DTT, 0% to 10% (v/v) DMSO, and different pH ranging from 6 to 10. The Km values for RNA and BBT-ATP substrates were determined in optimized buffer (50 mM Tris-Cl [pH 7.0], 0.01% Triton X-100, 1 mM MnCl2). The Km value for RNA was calculated with varying 3′UTR-U30 RNA concentration from 1.25 to 80 nM (at a fixed BBT-ATP concentration of 20 µM), whereas the BBT-ATP Km value was measured by varying the concentration from 0.3 to 20 µM in the reactions containing 80 nM of 3′UTR-U30 RNA. The Km app value for 3′UTR-U30 was determined in the presence of 2 µM BBT-ATP, and the Km app for BBT-ATP was obtained with a fixed concentration of 50 nM 3′-UTR-U30 RNA under similar assay conditions.
The enzyme stability was assessed using NS5 alone or by preincubation with either BBT-ATP or 3′UTR-U30 RNA in 15 µL of the optimized assay buffer for 0 to 4 h at room temperature. Reactions were started by adding 15 µL of the remaining substrates at 2× concentration. Reactions were quenched at 60 min with 20 µL of stop buffer containing CIP and further incubated for 1 h at room temperature to allow complete hydrolysis by the phosphatase. The release of the BBT was monitored as described.
Assay validation with reference compounds 3′dATP and adenosine nucleotide triphosphate analogues
The chain terminator 3′dATP and a triphosphorylated DENV NS5 RdRp nucleotide inhibitor (tpNITD008) 6 were used as reference compounds to validate the assay. Eight-point IC50 curves for values of these molecules were determined by 3-fold dilution from the highest inhibitor concentration of either 20 or 30 µM. These experiments were performed by dispensing 100× compound solution prepared in 90% (v/v) DMSO onto the plate, followed by adding NS5-RNA complex that had been preincubated for 30 min. Reactions were initiated by the addition of BBT-ATP solution. After a 60-min incubation at room temperature, the stop solution was added, and BBT production was monitored as described above.
Assay miniaturization and inhibitor screening
The assay was converted into a 384-well plate format with the reaction volume reduced to 10 µL. The reaction linearity, Z factor, S/N ratio, and IC50 of reference compounds were examined. After miniaturization, assay performance was evaluated in a pilot screen on a compound library. The screening flowchart is presented in
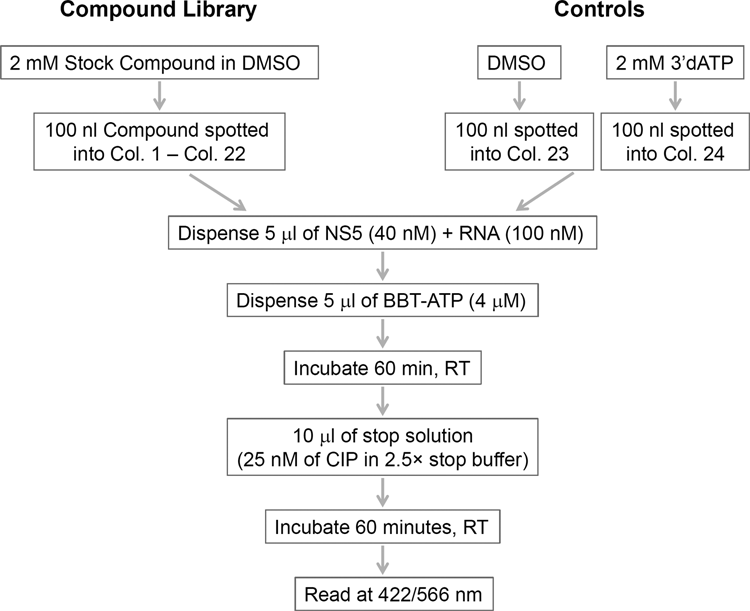
Screening workflow of the representative library in 384-well plates. CIP, calf intestinal alkaline phosphatase; RT, room temperature.
Data analysis
Km values were obtained by plotting the observed BBT production as a function of nucleotide or RNA concentrations, and the data were fitted to the equation v = Vmax {[substrate]/([substrate] + Km app)}. IC50 values were obtained by fitting the data to a 4-parameter logistic equation, Y = Bottom + (Top − Bottom)/(1 + (10^( logIC50 − X)^Hill slope)), where Bottom is the minimum Y value, Top is the maximum Y value, X is test compound concentration, and the Hill slope is the slope of the linear portion of the semi-log curve. IC50 value was extrapolated from logIC50 according to the GraphPad algorithm (GraphPad version 5; GraphPad Software, San Diego, CA). Z factor was calculated according to Zhang et al, 18 Z = 1 − ((3SDTot + 3SDMin)/(meanTot − meanMin)), and compound activity was calculated from the following: % inhibition = (100 − ((sample signal − meanMin)/(meanTot − meanMin) × 100)). Screening data were uploaded into ActivityBase (IDBS, Guildford, UK) equipped with the add-in XLfitR (V.5, IDBS) for data normalization and visualization in Microsoft Excel and Spotfire (TIPCO, London).
Results and Discussion
To perform a high-throughput in vitro enzymatic assay for DENV NS5 RdRp, we developed FAPA. The assay principle is depicted in
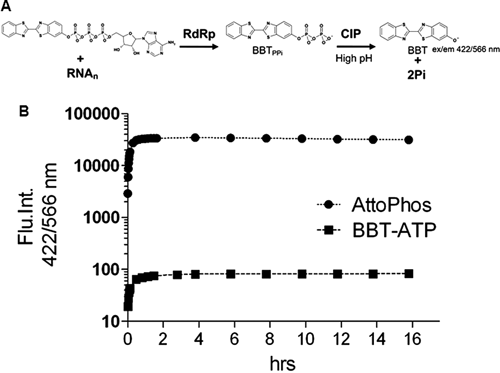
Fluorescence-based alkaline phosphatase–coupled polymerase assay (FAPA) principle. (
We chose the BBT fluorophore to develop an HTS assay on the basis of the following criteria: it has a high extinction coefficient, a large Stokes’ shift, and excellent photostability (ϵ = 26,484 M−1cm−1, exmax/emmax 422/566 nm). To examine BBT properties in terms of reactivity toward CIP hydrolysis, CIP reactions were optimized with AttoPhos® as the substrate mimic of the polymerase reaction by-product. Then, 5 µM AttoPhos® was treated with 0.01, 0.1, or 1 nM CIP in a total volume of 50 µL in a 96-well, half-well plate with NEB buffer no. 3 (100 mM NaCl, 10 mM MgCl2, and 1 mM DTT), with pH ranging from 7.5 to 10 (Tris-HCl [pH 7.5], HEPES-NaOH [pH 8.0-8.5], AMPSO-NaOH [pH 9], and CAPSO-NaOH [pH 9.5-10]). Liberation of BBT was monitored at 422/566 nm. AttoPhos® was well hydrolyzed by CIP (see
BBT calibration, detection limit, and assay optimization
To ensure that the fluorescence reading from AttoPhos®-CIP treatment truly reflects the RdRp activity, calibration values were compared between BBT and AttoPhos®-catalyzed CIP reactions. BBT or AttoPhos® at 2 to 200 nM was prepared in 30 µL RdRp test buffer (50 mM Tris-HCl [pH 7.0], 50 mM NaCl, 1 mM DTT, 1 mM MnCl2) before the addition of 20 µL of stop buffer containing 12.5 nM CIP (final CIP concentration at 5 nM in 50 µL). This 2-step procedure was performed to simulate the RdRp assay reaction. In addition, possible interference from the NS5 protein, RNA, and nucleotide triphosphates, which would eventually be present in the final reaction, was also investigated. CIP could hydrolyze most, if not all, the AttoPhos® in the test buffer (
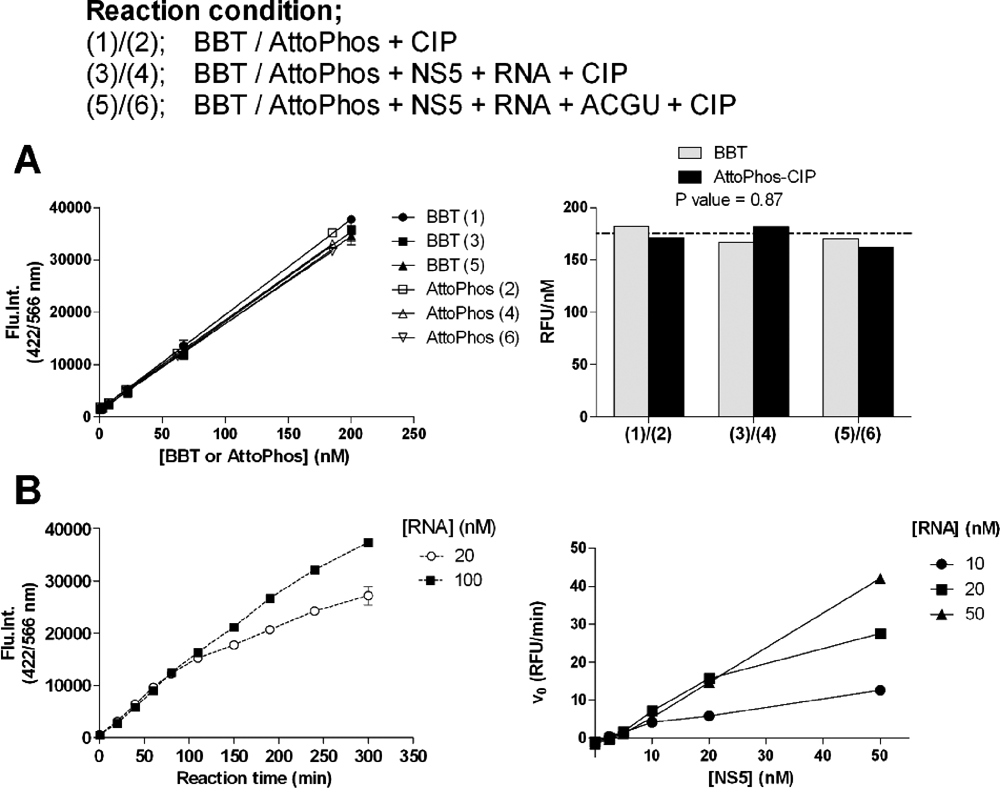
BBT standard curve and RNA-dependent RNA polymerase (RdRp) reaction. (
Assay optimization was performed in 30 µL in 96-well, half-well plates in the RdRp test buffer. We next evaluated if BBT-ATP was a substrate for the DENV NS5 RdRp. Reaction progress curves of 50 nM RdRp on either 20 nM or 100 nM 3′UTR-U30 with 1 µM BBT-ATP are shown in
FAPA for DENV-4 NS5 for HTS
RdRp activity in the optimized buffer was linear up to 120 min in a reaction that contained 20 nM NS5, 50 nM RNA, and 2 µM BBT-ATP (
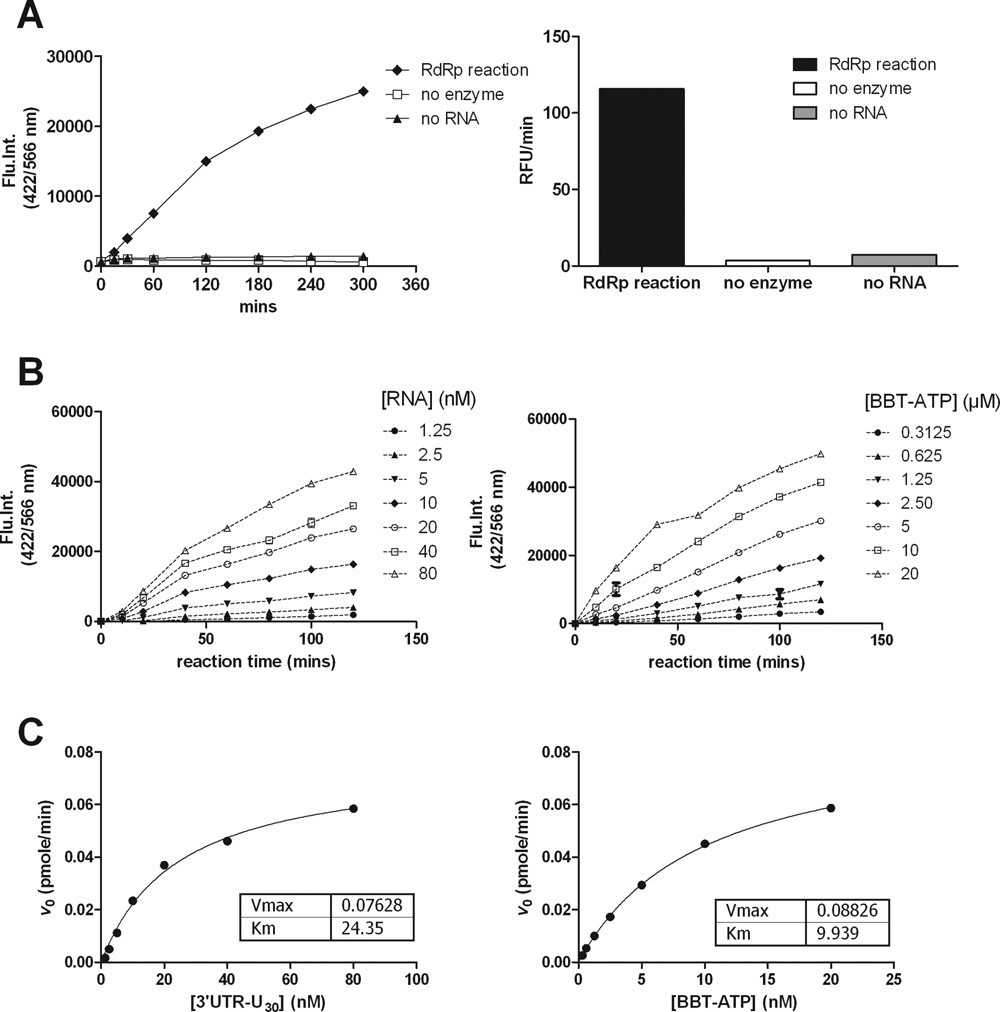
RNA-dependent RNA polymerase (RdRp) reaction kinetics. (
The RdRp reaction kinetics was determined for both the 3′UTR-U30 RNA and BBT-ATP. For the Km determinations, RNA concentrations ranging from 0 to 80 nM were assayed with a saturating BBT-ATP concentration of 20 µM or BBT-ATP concentrations ranging from 0.3 to 20 µM with 80 nM RNA. Reaction progress curves from 0 to 200 min were recorded to evaluate the reaction linearity (
The Km values for 3′UTR-U30 and BBT-ATP were 24 nM and 10 µM, respectively (
We next evaluated the stability of the assay reagents (see
Validation of FAPA
The reference compound 3′dATP and dengue adenosine nucleotide triphosphate analogue tpNITD008 were used to validate FAPA. Dose-response curves for 3′dATP and tpNITD008 are shown in
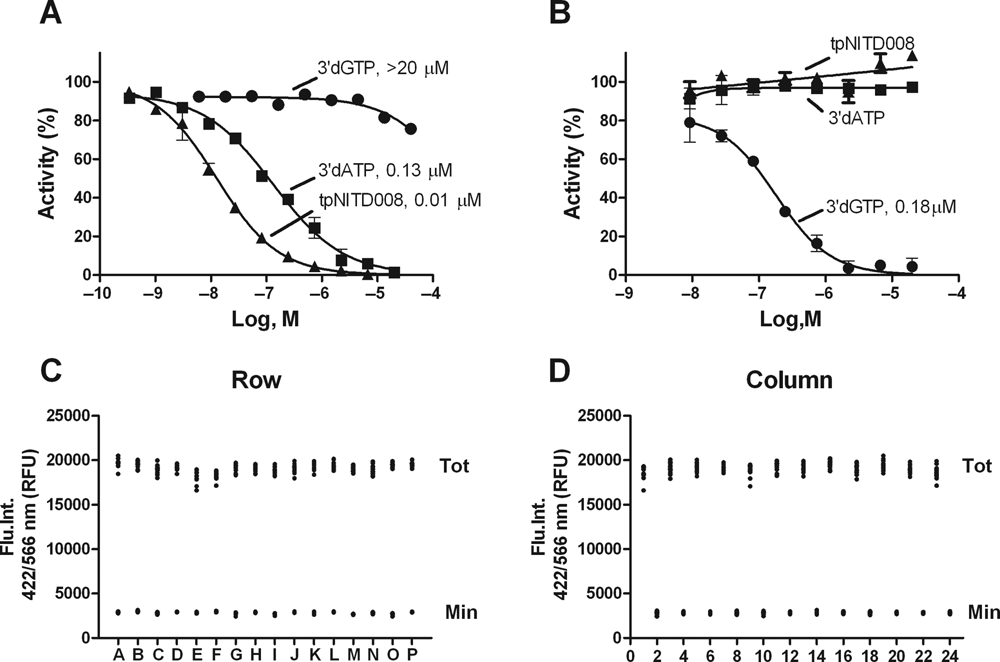
Assay validation, IC50 of reference compounds, and Z factor. (
Assay quality was assessed by Z factor determination in 384-well plates. The reaction volume was reduced to 10 µL for the RdRp reaction and 10 µL for the stop solution, keeping the final reagent concentrations the same. The plate uniformity was tested by spotting 100 nL of 90% (v/v) DMSO as vehicle controls onto even columns (“Tot” signal) and 100 nL of 2 mM of 3′dATP (final concentration 20 µM) onto odd columns (“Min” signal) across the whole plate (
Pilot screen with diverse compound library
A compound library of diverse structures selected from various vendors, comprising 40,572 compounds, was used in a pilot screen (118 plates in total, including 2 DMSO dummy plates). Screening (protocol is shown in
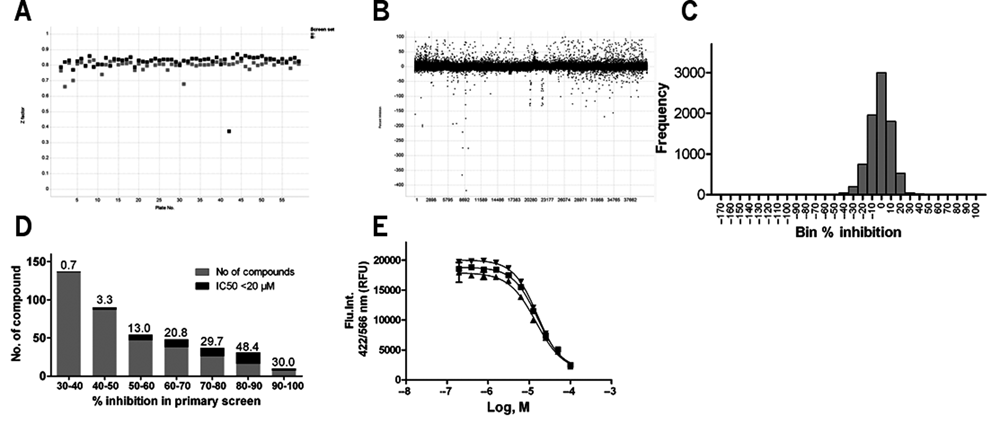
Compound library screening. (
In addition, NS5 from DENV serotypes 2 and 3 was included in this study to assess the adaptability of FAPA. To establish an assay for DENV-2 and DENV-3 NS5, we (1) determined the Km
app values for RNA and BBT-ATP, (2) determined the reaction linearity in a time course experiment, (3) obtained a Z factor of above 0.5, and (4) validated the assay with the 3′dATP reference compound. The kinetic parameters for DENV NS5 serotypes 2 and 3 and IC50 values for inhibitors are shown in
DENV-2, 3, 4 Kinetic Parameters and IC50 Values of the Reference Compounds
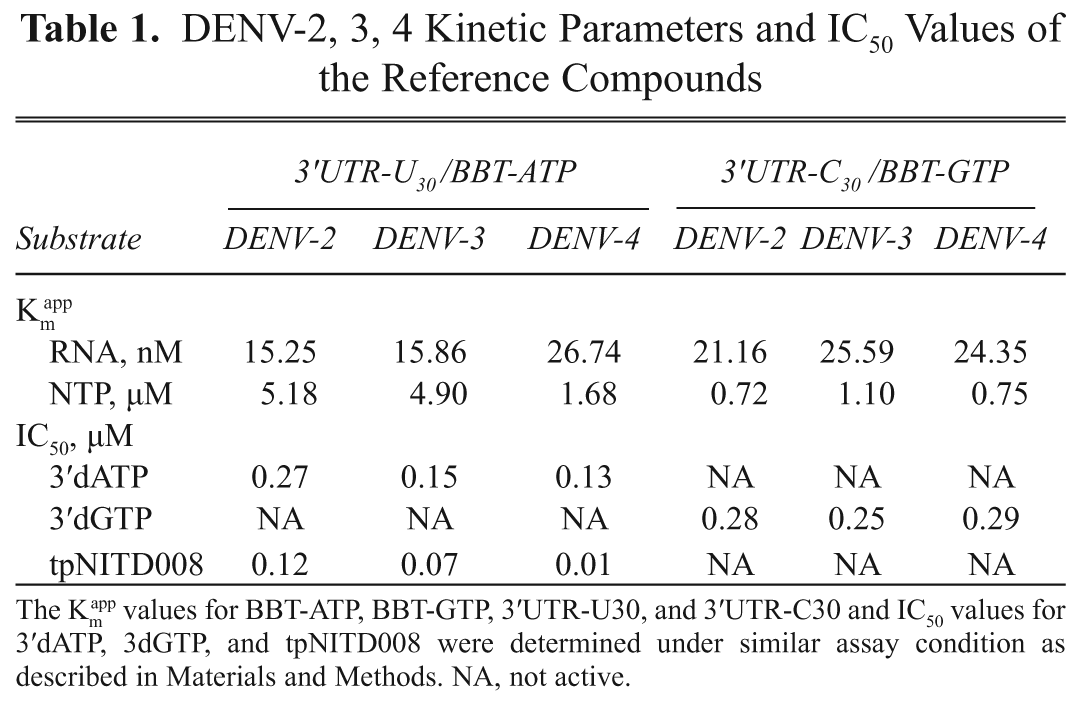
The Km app values for BBT-ATP, BBT-GTP, 3′UTR-U30, and 3′UTR-C30 and IC50 values for 3′dATP, 3dGTP, and tpNITD008 were determined under similar assay condition as described in Materials and Methods. NA, not active.
In summary, the FAPA is versatile, homogeneous, and robust. The method presented here should allow the screening of compounds targeting polymerases. The assay is robust enough to avoid identifying nonspecific inhibitors commonly detected in an HTS, facilitating the drug discovery process by identifying inhibitors that are specific to the polymerase protein target.
Footnotes
Acknowledgements
We thank Shamala Devi for providing RNA viruses for cloning of DENV-3 and DENV-4 NS5. In addition, we thank Shahul Nilar for compound selection, Amelia Yap and Wan Kah Fei for help with screening, and Christian Noble for help in manuscript editing.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.