
Abstract
A 2-step kinase assay was developed and used in a high-throughput screen (HTS) of more than 1 million compounds in an effort to identify c-Abl tyrosine kinase activators. This assay employed a 2-step phosphorylation reaction: in the first step, purified recombinant c-Abl was activated by incubating with compound in the presence of adenosine triphosphate (ATP). In the second step, the TAMRA-labeled IMAP Abltide substrate was added to allow phosphorylation of the substrate to occur. The assay was calibrated such that inactive c-Abl protein was activated by ATP alone to a degree that it not only demonstrated a measurable c-Abl activity but also maintained a robust assay window for screening. The screen resulted in 8624 primary hits with >30% response. Further analysis showed that 1024 had EC50 <10 µM with a max % response of >50%. These hits were structurally and chemically diverse with possibly different mechanisms for activating c-Abl. In addition, selective hits were shown to be cell permeable and were able to induce c-Abl activation as determined by In-Cell Western (ICW) analysis of HEK-MSRII cells transduced with BacMam virus expressing full-length c-Abl.
Introduction
D
Recently, there has been increased interest in identification and development of small-molecule kinase activators as a modality for alleviation of disease. 1 One example is AMP-activated protein kinase (AMPK), an important regulator of metabolic function. 2 Efforts have been made to discover and develop AMPK-specific small-molecule nonnucleoside activators of AMPK, 3-5 and the newly discovered thienopyridone A769662 validates the approach of AMPK activation as a way to ameliorate diabetes symptoms by inhibiting lipogenesis 6 and increasing glucose uptake 7 in cellular and animal models.
c-Abl, a member of the Src family of nonreceptor tyrosine kinases ubiquitously expressed in mammalian cells, is found throughout the cell, including the nucleus, cytoplasm, mitochondria, endoplasmic reticulum, and plasma membrane. 8-11 c-Abl is expressed in 2 isoforms (1a and 1b) in humans as a result of alternative mRNA splicing of the first exon. 12 The 1b form is myristoylated at the N-terminus due to posttranslational modification, whereas the 1a form is 19 residues shorter at the N-terminus and not known to be myristoylated. The N-terminal half of c-Abl consists of an SH3 domain, an SH2 domain, and a catalytic kinase domain. The C-terminal half of c-Abl contains a DNA binding domain, an actin binding domain, and various short recognition motifs, including PxxP. 13
Through interaction with a large variety of cellular proteins such as signaling adaptors, kinases, phosphatases, transcription factors, and cytoskeletal proteins, c-Abl influences a diverse range of cellular processes, with its role in hematopoesis perhaps best characterized. Gene knockout experiments in mice have shown that abl proteins play important roles in cell homoeostasis. 14,15 Although the role of nuclear c-Abl has been well explored, 13 the role of cytoplasmic c-Abl deserves further study.
Under normal conditions, c-Abl adopts a closed, inactive conformation in the cell. Recent X-ray crystal structures of the truncated 1b form (1-531) 16 and N-myristoyl-cap deleted form (with residues 15-56 deleted) 17 shed light on how the kinase activity is regulated. These studies show that the N-terminal myristoyl cap binds to the base of the kinase domain and induces conformational changes, allowing the rigid SH2 and SH3 domains to dock onto it and restrict c-Abl to a compact inactive conformation. By deleting amino acid residues 15 to 56 in the N-cap of c-Abl, Nagar et al. 17 further demonstrated that there are 3 critical linchpins for maintaining the compact state: the docking of the SH2 domain, the docking of the SH3 domain to the polyproline helix formed by the SH2-kinase linker, and the brace formed by the N-terminal myristoyl cap. In addition, a phosphoserine residue in the N-cap (pSer69) interacts with the SH3-SH2 connector, thereby restricting movement of the SH3-SH2 domain.
c-Abl is also regulated by phosphorylation. Twelve phosphorylation sites on tyrosine residues have been identified in the N-terminus of c-Abl, and the phosphorylation of Y245 and Y412 is required for c-Abl activation. 18 The involvement of other kinases in regulating c-Abl activity remains to be determined.
Based on the crystal structure data, a mechanism of c-Abl activation was proposed that involves opening the closed confirmation and phosphorylation of Y245 in the linker and Y412 in the activation loop. 19 There are numerous experimental data in support of this proposed mechanism. Studies show that phosphopeptide binding to the src homology (SH) SH2 domain and mutations of the myristoylation site 19 are able to activate c-Abl. Mutations around the SH3-SH2 domains and the SH2/kinase domain linker region are also able to disrupt the closed confirmation and thereby activate c-Abl. 18,20,21 More recently, a potent and highly specific FN3 monobody has been shown to disrupt the interaction of SH2 domain and kinase domain and therefore is able to activate the kinase in vitro. 22 Another mechanism for c-Abl activation is demonstrated by activation of c-Abl by binding of a peptide with micromolar affinity for the SH3 domain. 23 In addition, N-terminal deletion through gene fusion, as demonstrated by the BCR-ABL protein, produces a mutant protein that exhibits constitutive high activity and aberrant signaling, 24 which is the primary causative factor of chronic myelogenous leukemia (CML).
Although c-Abl can be activated through different mechanisms, the common theme seems to be the opening up of the closed, autoinhibited conformation followed by phosphorylation. Upon activation, c-Abl relaxes its compact conformation and adopts an elongated shape with the SH2 domain residing on top of the N-lobe of the kinase domain and the SH3 domain dissociated from the C-lobe of the kinase domain. 17 The resultant exposure of the linker and the activation loop to phosphorylation thereby determines the degree of activation of c-Abl and therefore its catalytic activity.
Recent studies have shown that c-Abl is an essential regulator of normal morphology of mammary epithelial cells (MECs) and is a suppressor of oncogenic transforming growth factor-β (TGF-β) signaling during mammary tumorigenesis. c-Abl inactivation was sufficient to induce phenotypic and transcriptional epithelial mesenchymal transition in normal MECs. Conversely, c-Abl activation restores normal MEC morphology and TGF-β-mediated cytostasis in metastatic MECs and blocks their secretion of matrix metalloproteinases (MMP-3 and -9) induced by TGF-β. 25 In addition, c-Abl activation prevents 4T1 tumor growth in mice. Consistent with these results, clinical trials with c-Abl inhibitor imatinib found that c-Abl ablation enhances the tumorigenicity of breast cancer cells. 26 Therefore, c-Abl activators could potentially inhibit breast cancer development and progression.
We describe here a novel approach to identify c-Abl activators using an IMAP assay. IMAP (http://www.moleculardevices.com/pages/reagents/imap_intro.html) is a technology based on the specific and high-affinity interaction of trivalent metal containing nanoparticles with phosphogroups. The capture of phosphorylated, fluorescently labeled peptide in a kinase reaction gives rise to a higher fluorescent polarization (FP) or time-resolved fluorescence resonance energy transfer (TR-FRET) signal, which directly measures kinase activity. First, we generated a recombinant c-Abl construct (1-531) and expressed the protein in the presence of imatinib to constrain the protein to adopt an inactive conformation. We then calibrated the assay conditions to maximize the chance of identifying potential c-Abl activators. After activation, the enzyme activity was measured using Abltide substrate in the IMAP format. More than 1 million compounds were screened at 10 µM, and hundreds of hits with sub-micromolar potency were identified and confirmed in an orthogonal assay. In addition, the cellular potency of compounds was measured in a cell-based assay as a precursor to further in vitro and in vivo phenotypic endpoint assays.
Materials and Methods
Cloning, expression, and purification of c-Abl
The full-length Homo sapiens ABL1b was generated from human CML Marathon cDNA (Clontech, Mountain View, CA) and cloned into the GateWay pENTR/Tev/D-TOPO vector (Invitrogen, Carlsbad, CA). c-Abl(1-531)-His6 fusion (c-Abl) was generated by standard PCR technique using primers designed to incorporate both Kozak sequence CACCATG to the 5′-end and an in-frame hexahistidine tag sequence to the 3′-end of the coding sequence. The PCR product was first cloned into a Gateway pENTR vector and then transferred into GateWay pDEST8 baculovirus transfer vector by LR reaction as described by the manufacturer (Invitrogen). Recombinant baculovirus was generated as described in the Bac-2-Bac manual (Invitrogen). Both P1 virus and baculovirus-infected insect cells (BIICs) were generated. 27 Recombinant protein expression was achieved by infecting 1 × 106/mL of exponentially growing Spodoptera frugiperda cells with a 10,000-fold diluted BIIC stock. Imatinib (Gleevec) was then added to the final concentration of 10 µM to generate c-Abl/imatinib complex during protein expression. The infection was allowed to proceed for the duration of 72 h at 27°C before the culture was harvested by centrifugation. Cell pellets were lysed by sonication in Buffer A (50 mM Tris-HCl [pH 8.0], 0.6 M NaCl, 5 mM B-ME, 1% Triton-X-100) plus Complete Proteinase Inhibitor (Roche Diagnostics, Nutley, NJ) followed by centrifugation. The supernatant was bound to Ni-NTA Superflow resin (Qiagen, Germantown, MD), washed with Buffer A + 40 mM imidazole, and protein was eluted with Buffer A + 500 mM imidazole. Eluate was dialyzed against Buffer B (50 mM Tris-HCl [pH 7.5], 0.25 M NaCl, 3 mM dithiothreitol [DTT], 5% glycerol) followed by centrifugation to remove insoluble material.
Protein QC
Purified c-Abl protein (1-531) was quantified by the measurement of absorbance at A280 nm and then loaded at a range of protein concentrations onto 10% NU-PAGE gels with SeeBlue Plus2 prestained standard and run in MES buffer according to the manufacturer’s protocol (Invitrogen). Gels were stained with SimplyBlue SafeStain (Invitrogen) according to the manufacturer’s protocol and scanned on Kodak Image Station 4000 mm and bands analyzed using Kodak Molecular Image Software 4.0.5 (Eastman Kodak, Rochester, NY).
Mass spectrometry analysis of imatinib concentration in protein
A 2-µL c-Abl protein sample was injected onto an ACQUITY UPLC BEH (2.1 × 50 mm, 1.7 µm, C18) column connected to an ACQUITY PDA and a Waters SQD mass spectrometer (LC/MS) equipped with an electrospray ionization source (Waters Corporation, Milford, MA). The mobile phase consisted of solvent A: 0.1% formic acid and solvent B: 0.1% formic acid in CH3CN at a flow rate of 1.0 mL/min. Imatinib was eluted using a linear gradient of 3% to 100% B spanning 2 min. Using positive ion detection, the protonated molecular ion (M+H)+ m/z 494 along with 2 fragment ions m/z 268 and m/z 248 were used to identify the elution of imatinib. The LC/MS system was controlled and the data were processed using Waters’ MassLynx and QuantLynx 4.1 software packages, respectively.
Filter binding experiment of c-Abl activation assay
The reactions were carried out in 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS; pH 7.2), 2 mM MgCl2, 1 mM DTT, 0.1 mg/mL bovine serum albumin (BSA), 0.01% Tween-20, and 25 mM NaCl in a 96-well NBS (nonbinding surface) plate (Costar, #3884). c-Abl activation was monitored in a 2-step reaction. In the first step, c-Abl was allowed to undergo autoactivation. A grid titration experiment was performed by varying c-Abl concentration (2.5-10 nM final) and adenosine triphosphate (ATP) concentration (6.25-100 µM final). The autoactivation reaction was allowed at room temperature for 20 min in a total reaction volume of 40 µL. In the second step, the amount of c-Abl kinase activity generated in the first activation step was measured in a kinase activity assay with Abltide, EAIYAAPFAKKK. In this step, 10 µL of the activation mix described above was transferred to 40 µL of the substrate mix containing Abltide and 33P-ATP at final concentrations of 11.5 µM Abltide of desired c-Abl and ATP concentrations. The reaction was run at room temperature for 10 min and quenched by the addition of 50 µL/well of 1% phosphoric acid solution. The quenched reactions were transferred to a 96-well MultiScreen-PH filter plate (MAPHN0B50; Millipore, Billerica, MA) which was washed, dried, and counted for radioactivity incorporation as described previously. The amount of product (33P-labeled Abltide) formation was measured.
Preparation of compound plates for high-throughput screening and profiling
To each well of black low-volume 384-well plates (cat. no. 782076; Greiner Bio-One, Monroe, NC), 100 nL of compound in DMSO was added using Cartesian Hummingbird (Genomics Solutions, Ann Arbor, MI). Compounds assessed in full-curve were serial diluted by BioMekFX (Beckman Coulter, Fullerton, CA) prior to nanoliter dispensing by Hummingbird. For In-Cell Western assays, 1 µL of compound in DMSO was added to clear V-bottom polypropylene 384-well plates (Greiner Bio-One) by Biomek FX.
IMAP assays
All IMAP reactions were carried out in IMAP Reaction Buffer (10 mM Tris [pH 7.2], 10 mM MgCl2, 25 mM NaCl, 0.01% Tween-20) at 100 nM final concentration of IMAP Abltide (5′TAMRA-KKGEAIYAAPFA-NH2) followed by quenching and binding with the addition of IMAP Binding Buffer (1:200 of IMAP Progressive Binding Reagent in 60% IMAP Progressive Binding Buffer A/40% IMAP Progressive Binding Buffer B), which was obtained from Molecular Devices (now MDS, Sunnyvale, CA).
Reagents for calibrating the assay were generated as follows: 2 µM c-Abl protein was incubated for 2 h at room temperature (RT) +/-500 µM ATP in IMAP Reaction Buffer (addition of 500 µM ATP fully activates c-Abl). Samples were then snap-frozen in liquid nitrogen for subsequent assays. The linearity of IMAP reaction time was determined by titrating fully activated c-Abl and stopping the IMAP reaction at a range of time points. Percent activation was determined with a 3-way grid experiment to determine rate (V/[E], (mP/min)/nM) of c-Abl at a range of concentrations of c-Abl and ATP at a range of activation times followed by 15-min IMAP Abltide phosphorylation reactions. V/[E] was also determined for fully activated c-Abl, and the average percent activity of c-Abl in the grid experiment was determined as a percentage of the V/[E] of fully activated c-Abl.
High-throughput screening (HTS) and dose-response confirmation assays were read on EnVision (PerkinElmer, Waltham, MA) with a TAMRA FP program (531 ex, 595 em, BODIPY-TMR dual [561 dichroic]). DMSO tolerance, reagent stability, and signal stability optimization experiments were read on Acquest plate readers (Molecular Devices, now MDS).
For the IMAP HTS campaign, 1 µL of 125 nM c-Abl and 1 µL of 100 µM ATP were added to each sample well of compound plates. Then, 1 µL of 125 nM c-Abl and 1 µL of 900 µM ATP were added to high control wells. These additions were followed by a 30-s spin at 1000 rpm in centrifuge. At 10 min post-ATP addition, 8 µL of 125 nM IMAP peptide was added to each well. After a 15-min incubation with IMAP peptide, this reaction was terminated by addition of Binding Buffer. Plates were covered and incubated 1 h, followed by a spin at 1000 rpm in a centrifuge, and then the plates were read on EnVision. For dose-response confirmation, the conditions were the same as the above HTS conditions, except 1 µL of 300 µM ATP was added to all sample wells instead of 100 µM ATP. Compounds were plated in 11-point curves diluted 1:3 from 50 µM top concentrations (final assay concentration) and 1% DMSO.
To triage autofluorescent compounds, total fluorescence intensity (FI) was calculated for each compound identified as a hit in HTS as follows: Total FI = parallel intensity + 2 * perpendicular intensity. Compounds that had total Total FI >150% of controls or <50% of controls were excluded. In addition, fluorescence pXC50 was determined concomitantly with IMAP pEC50 of these compounds.
In-Cell Western
A 384-well (In-Cell Western [ICW]) assay was established using HEK-293MSRII cells transduced with a BacMam virus 28 expressing full-length c-Abl and plated in DME/F12 + 0.1% fetal bovine serum (FBS; Invitrogen) followed by O/N incubation at 37°C, 5% CO2. Compounds were added to cells by a Cybi-Well Dispenser (Cybio, Woburn, MA). After a 5-min incubation, cells were prepared for ICW as detailed in the LICOR ICW Kit II (biosupport.licor.com/docs/ICW_Assay_Kit_08911.pdf) with antibody/counterstain concentrations as follows: 1:500 dilution of anti-phospho-c-Abl (pTyr245) antibody (Sigma, St. Louis, MO), 1:1200 dilution of goat-antirabbit IRDye800 secondary antibody, 1:1000 dilution of Sapphire 700, and 500 nM Draq 5 (Li-COR, Lincoln, NE). All ICW additions and washes following compound treatment were performed on a CRS platform outfitted with multiple dispensers and a Tecan plate washer (Tecan, Männedorf, Switzerland). Plates were then read on the Aerius Plate reader (Li-COR) according to the manufacturer’s standard protocols.
Data Processing and Analysis
Robust Z′ 29 and pXC50 values were determined using the XC50 module within Abase (IDBS, Guilford, Surrey, UK). IMAP HTS data were collected from plate readers, and percent response (% Response) was determined for each point as follows: % Response = 100 * (FP High Controlavg – FP Sample)/(FP High Controlavg – FP Low Controlavg). Data were visualized within Spotfire (Tibco Software, Palo Alto, CA), GraphPad Prism (GraphPad Software, La Jolla, CA), and KaleidaGraph 4.1 (Synergy Software, Reading, PA).
Results
Protein quality control
Purified c-Abl protein was detected as a ~60-kDa major band with >90% purity by densitometry on the Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel ( Fig. 1A ). This is consistent with the expected molecular weight (MW) of c-Abl (1-531) that contains the N-terminal myristoyl group. Only at the highest protein concentration (10 µg) could minor contaminating bands be detected on the gel. LC/MS analysis revealed a series of peaks at 61,035, 61,117, 61,198, 61,279, 61,358, and 61,439 Da ( Fig. 1B ). These peaks are separated by a distance of ca. 80 Da, which is attributable to differences in the phosphorylation state of the protein sample. These phosphorylation sites are on neither Y245 nor Y412 residues (data not shown). This is in agreement with the activity data shown in Figure 3A that, prior to activation, purified c-Abl is inactive (unable to phosphorylate Abltide) as phosphorylation on Y245 and Y412 is essential to the enzyme activity.
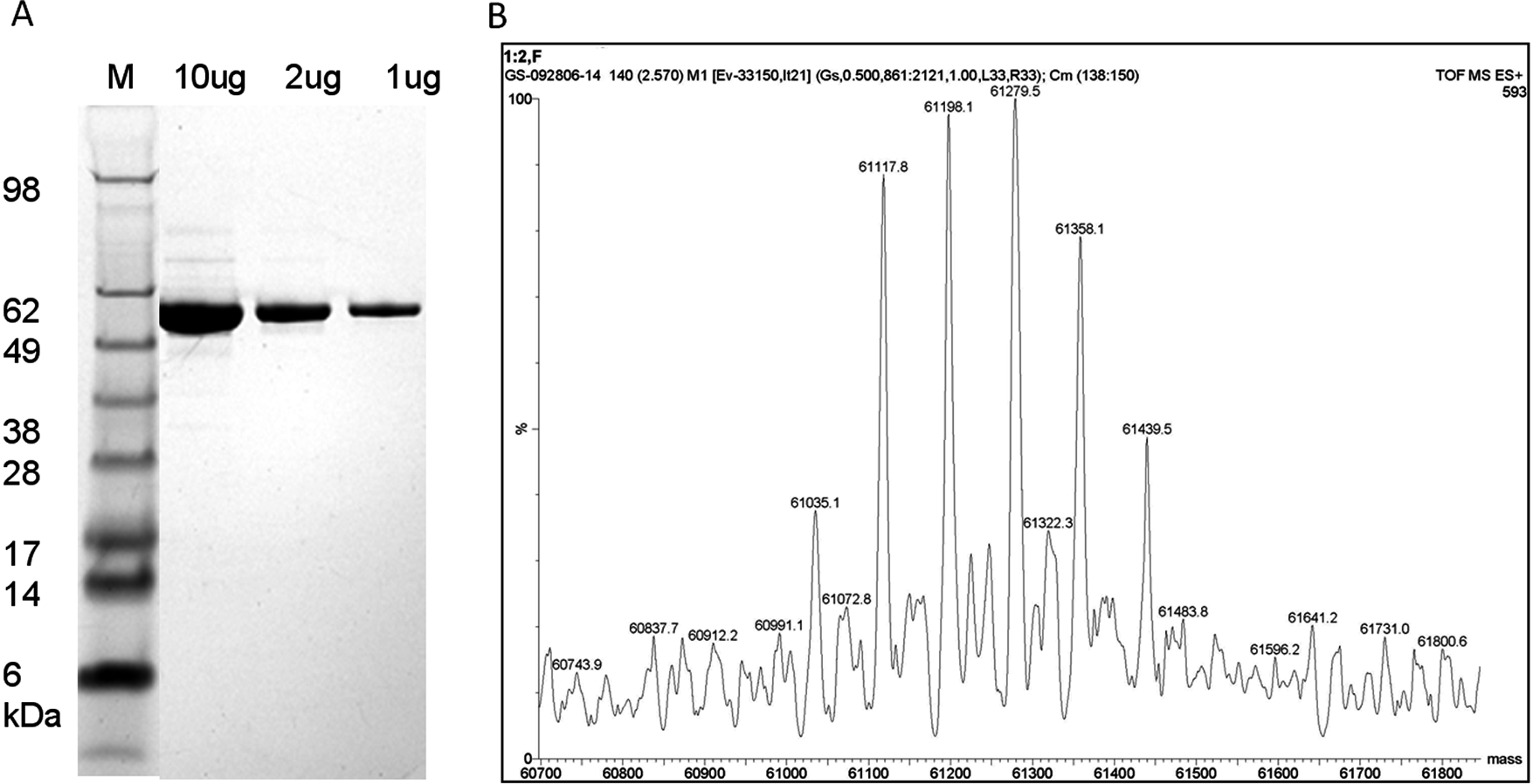
Quality control analysis of purified c-Abl (1-531). (
Confirmation that recombinant c-Abl is activatable
Samples of the purified c-Abl protein were tested in the filtration binding assay to affirm that the protein is inactive but activatable upon exposure to sufficient quantities of ATP. At 5 nM c-Abl, there was a clear increase in Abltide phosphorylation dependent on ATP concentration, with the activation of the c-Abl appearing to peak by ~60 min at 100 µM ATP ( Fig. 2A ). At 25 µM ATP, Abltide phosphorylation was dose dependent on c-Abl addition, but 10 nM c-Abl incubated with the 25 µM ATP was still not fully active when compared to 5 nM c-Abl, which had been fully activated prior to exposure to Abltide and 33P-ATP ( Fig. 2B ).
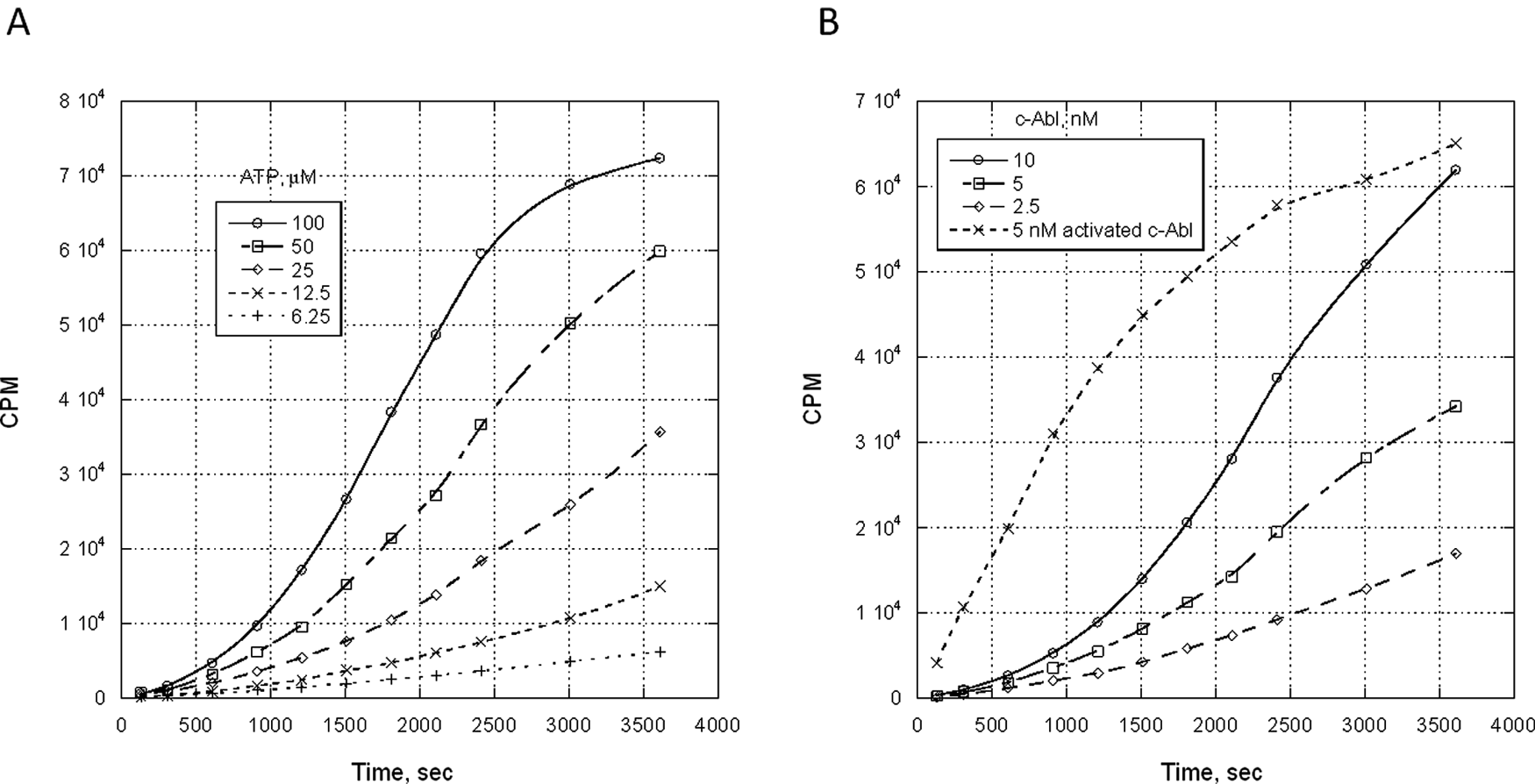
Activation profile of purified c-Abl protein. (
Calibration of IMAP activation assay
To determine the linearity of the activation step of the IMAP assay, a single pot of c-Abl activation reaction mixture was made containing 2 µM c-Abl enzyme and 500 µM ATP. Aliquots of this reaction were quenched with 50 mM EDTA at times shown in Figure 3A . These aliquots were then diluted and assayed at 1.25 nM c-Abl, 100 µM ATP, and 100 nM IMAP-Abltide in the assay buffer as described in the Materials and Methods section. This experiment showed that activation proceeded in a linear fashion up to 15 min ( Fig. 3A ). A grid titration experiment showed that phosphorylation of IMAP-Abltide by fully activated c-Abl was linear up to 20 min at up to 10 nM c-Abl ( Fig. 3B ). We tailored further optimization reactions to these limits to stay in the linear range of the assay.
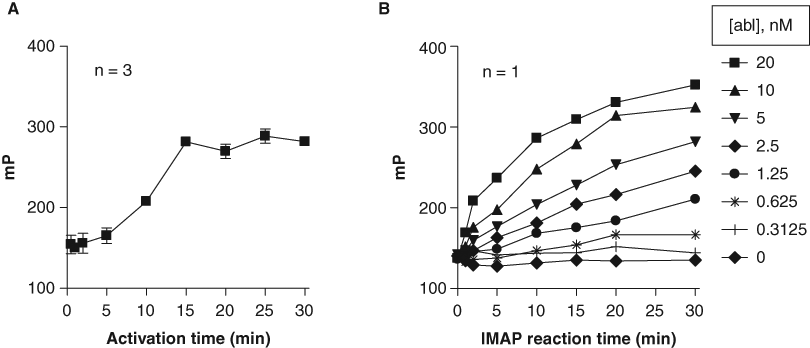
Calibration of c-Abl activation and substrate phosphorylation reactions in the IMAP assay. (
To detect potentially weak c-Abl activators in the compound collection, we calibrated the concentration of ATP in the assay to allow a basal level of c-Abl phosphorylation-dependent activation of ≤10%. To optimize this, we measured the initial velocity (V) of product formation (mP/min) at various fully activated c-Abl enzyme concentrations ([E]) in the presence of 30 µM ATP and determined average V/[E] = 4.39 ( Table 1 ). Next, we determined V/[E] in a grid experiment titrating inactive c-Abl against ATP incubated for 10 min prior to addition of substrate. V/[E] was determined at each set of conditions, and % activation was determined as a percent of the fully activated enzyme average determined above (V/[E] = 4.39 mP/min/nM). All conditions generated % activation of <10% ( Table 2 ). Out of these conditions, we selected a condition that would produce >80 mP assay window and robust Z′ > 0.6 to be used in HTS. A flowchart of the final assay conditions is shown in Figure 4 , and reagent conditions are as follows: 12.5 nM Abl, 10 µM ATP, 10-min activation time at ambient temperature, and 30-min IMAP reaction time with 100 nM IMAP-Abltide.
Initial Velocity Normalized against Enzyme Concentration
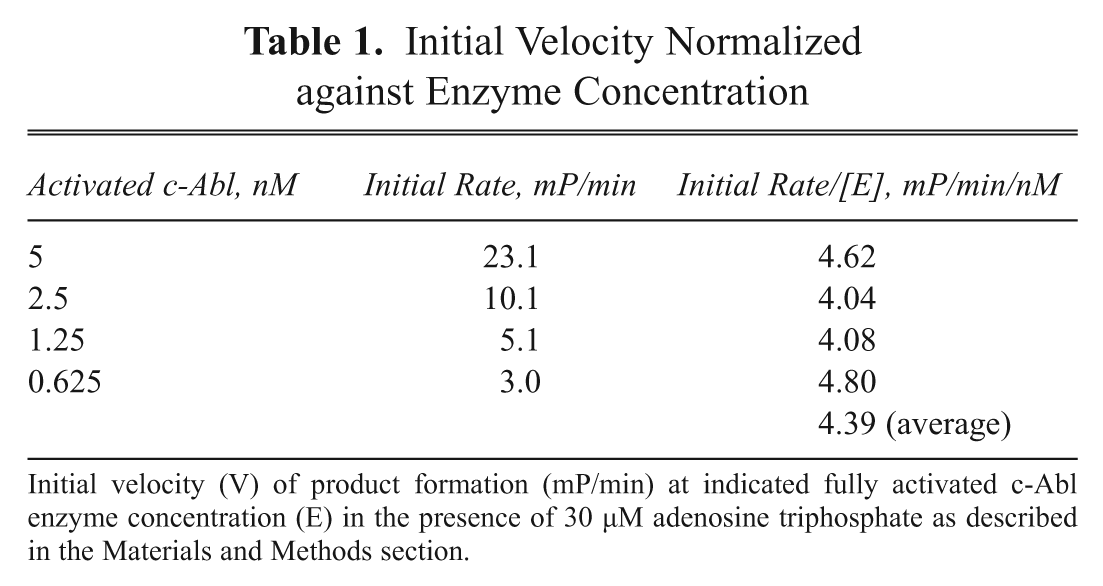
Initial velocity (V) of product formation (mP/min) at indicated fully activated c-Abl enzyme concentration (E) in the presence of 30 µM adenosine triphosphate as described in the Materials and Methods section.
Percentage Activation of Inactive c-Abl
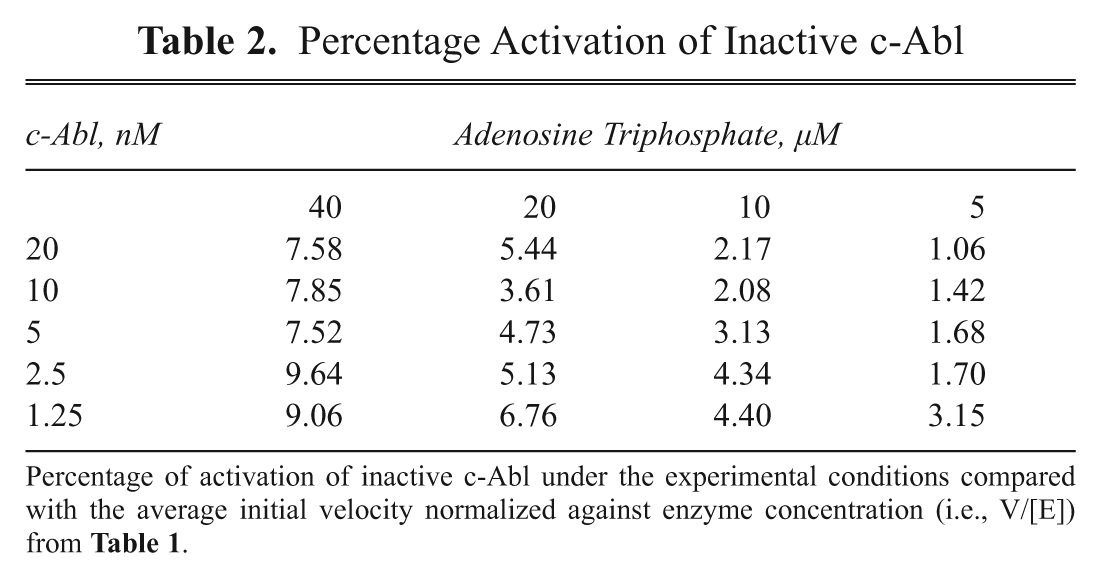
Percentage of activation of inactive c-Abl under the experimental conditions compared with the average initial velocity normalized against enzyme concentration (i.e., V/[E]) from Table 1 .
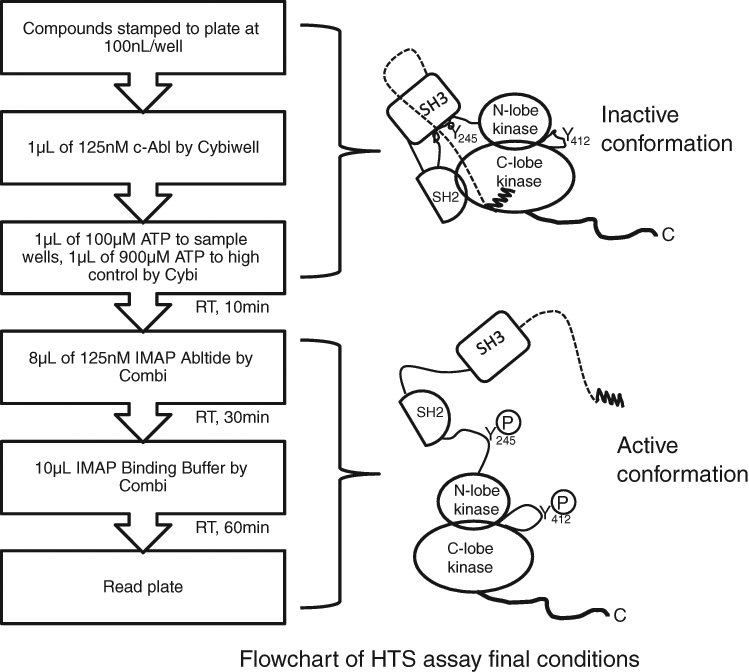
Flowchart of 2-step c-Abl kinase assay developed for high-throughput screening. Left panel: assay logistics; right panel: c-Abl conformation change before and after activation. Before activation, c-Abl adopts a compact (closed) conformation. After activation, c-Abl adopts an elongated conformation with the SH2 domain residing on top of the N-lobe of the kinase domain and the SH3 domain dissociated from the C-lobe of the kinase domain. Also shown are 2 key tyrosine residues, Y245 and Y412, which are phosphorylated upon activation.
Optimization of IMAP conditions for HTS
To test the suitability of the assay for HTS, we determined DMSO sensitivity, reagent stability, and signal stability as described below. The assay was first determined to be tolerant of DMSO from 0% to 10% in the activation step (0%-2% final). Although the average FP value of high control and mP window decreased slightly between 2.5% and 10% of DMSO, the robust Z′ was maintained at above 0.6 at all DMSO concentrations (data not shown). Assay reagents were stable at 4°C or RT for up to 7 h as demonstrated by maintenance of mP values and robust Z′, although mP values decreased modestly at all times >4 h under both conditions (data not shown). A plate of low and high control wells was set up with the same conditions as HTS (1% DMSO final) and read repeatedly at a 1-h interval for 18 h. It showed that mP window and robust Z′ values were stable for 12 h after the first read. However, the robust Z′ degraded after ~12 h, which was likely due to evaporation of samples in the assay plate while the assay plate resided in the reader (data not shown).
Outcome of primary HTS
In total, 1,333,024 compounds were screened at 50 µM in the first activation step (10 µM final) in 12 working days with a 2.5% plate failure rate and an average Z′ of 0.72 (data not shown). Using a cutoff of 30% activation, 8624 compounds were selected for confirmation at 50 µM in duplicate. Of these, 5392 compounds demonstrated at least 25% activation in at least 1 test set. We then applied activity-weighted diversity selection on these confirmed hits to select 4000 for full-curve confirmation. Hits were clustered and then prioritized based on the most potent compounds within each cluster. These 4000 compounds were tested in duplicate in 11 doses starting from 250 µM in the activation step as described in the Materials and Methods section.
Results of dose-response confirmation
The potencies, expressed as pEC50s, of the 4000 compounds selected for full-curve analysis ranged from 4 to 7.9. For follow-up studies in complementary biochemical and cellular c-Abl binding and activation assays, we arbitrarily removed compounds with total fluorescence intensity values greater than 150% or less than 75% of the control wells (i.e., DMSO), which were regarded as interfering compounds ( Fig. 5A ). Of these 4000 compounds, 2095 had an average pEC50 >5.0, 223 had an average pEC50 >6.0, and 3 had an average pEC50 >7.0.
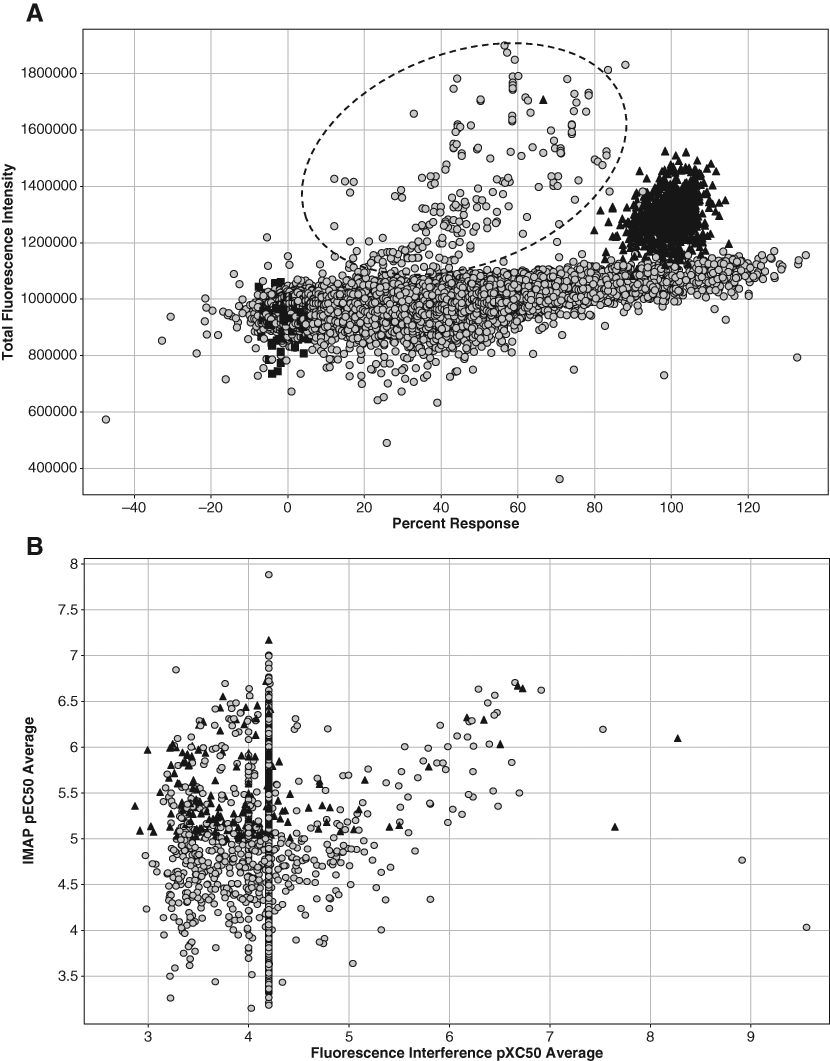
Spotfire plots of high-throughput screening data. (
Unlike in an enzyme inhibition assay, we reported 2 parameters for this enzyme activation assay. One parameter is the potency of the hit, expressed as pEC50, and the other is maximal percentage of activation normalized against the 2 controls (high and low) in the assay. To further narrow the list down to a manageable number, we selected only those compounds that had IMAP pEC50 >5.0 in at least 1 replicate and a maximum % response greater than 50%. A total of 1664 compounds fit these criteria. Looking over the full curve data for the compounds not selected, we rescued 2 compounds that had initially been eliminated, bringing the list to 1666 compounds. The list was further narrowed to 1024 compounds (32 plates, our arbitrary limit for follow-up) after removing compounds with pXC50 >5.0 in the fluorescence interference assay and allowing the exception of a dozen or so compounds with appealing chemical structures ( Fig. 5B ).
Further analysis of these 1024 hits showed that they belong to more than 10 chemotypes, exemplified by indoles, pyrazoles, and acylamino thiazoles.
ICW analysis of HTS hits
ICW analysis was used to confirm cellular activity of the c-Abl activators identified by IMAP assay in the HTS. The ICW assay offers a medium-throughput method for compound profiling prior to selection of potent hits for cellular mechanistic studies and in vivo proof-of-concept experiments. In this experiment, phosphorylated c-Abl (Y245) antibody along with a fluorophore-labeled species-specific secondary antibody was used. The fluorescence intensity of the antibody staining is then divided by the intensity produced by a mixture of nuclear counterstains to normalize the data against cell number. This assay detects c-Abl phosphorylation at Y245, which we determined to be necessary for c-Abl kinase activity in a variety of in vitro kinase assays (data not shown). A sample of phospho-c-Abl (Y245) ICW data from 5 compounds identified by HTS is shown in Figure 6 . A notable decrease in potency of ~1 log unit is observed between potency in the IMAP assay and the ICW assay. In addition, we observed that these compounds demonstrated a range of pEC50 values while eliciting a range of maximum % responses in both the IMAP and the ICW assays.
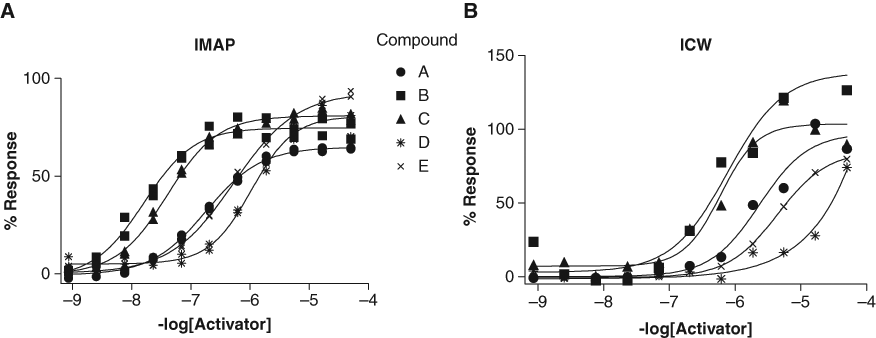
Comparison of 11-point pEC50 curves of 5 sample compounds run at half-log serial dilutions from 50-mM final assay concentration. (
Discussion
The goal of the current study is to identify small-molecule, potent c-Abl tyrosine kinase activators, so the critical component is a highly purified c-Abl protein in an inactive conformation that can be readily activated. To keep c-Abl stabilized in this compact state, c-Abl (1-531) was expressed in the presence of 10 µM imatinib. The protein shows >90% purity ( Fig. 1A ) and expected molecular weight for the 1-531 construct (including the myristoyl moiety) ( Fig. 1B ). The first question we asked was whether the protein was activatable. In the absence of ATP treatment, there was no phosphorylation on Y412, and the kinase activity was very low (data not shown). However, in the presence of ATP, the kinase activity showed ~10-fold increase, and % phosphorylation of Y412 increased from 0% to ~60% (data not shown). On the contrary, c-Abl protein that was expressed in the absence of imatinib was fully active before ATP incubation and had significant phosphorylation on Y412 (data not shown). These data are in agreement with the report that c-Abl autophosphorylates Y412 in the A-loop in the presence of ATP by a transmechanism. 30 Subsequently, a filter binding experiment was carried out to measure the effect of ATP or c-Abl concentrations on the rate of c-Abl activation. Toward this end, purified c-Abl protein was incubated either at fixed c-Abl concentration with varied ATP concentration ( Fig. 2A ) or at fixed ATP concentration with varied c-Abl enzyme concentrations ( Fig. 2B ). In both cases, the amount of phosphorylated Abltide was shown to be dose dependent to either ATP concentration or c-Abl concentration. The nonlinear progress curve further affirmed c-Abl activation as the reaction proceeded ( Fig. 2B ).
A sensitive and robust assay is critical to ensure a successful HTS campaign. A search for kinase activators also requires fine-tuning an assay to maximize the chance of finding all possible activators. Thus, we had the following criteria in mind for the assay: (1) sensitivity, (2) robustness, and (3) properly calibrated basal activation. As shown in Figure 3A , the c-Abl activation time is linear up to 15 min under the experimental conditions used. To measure the assay sensitivity, we performed a grid titration experiment ( Fig. 3B ) where we looked at signal linearity against IMAP reaction time and c-Abl concentration using preactivated c-Abl. It clearly demonstrated that a condition could be chosen from the grid titration experiment to ensure assay sensitivity and robustness.
To maximize the chance of identifying potentially weak c-Abl activators in the HTS, we deliberately calibrated the assay conditions (e.g., ATP and c-Abl concentrations and the total incubation time) to allow a basal level of phosphorylation-dependent activation of no more than 10% prior to compound addition. This partial-activation approach is frequently used for identifying G-protein-coupled receptor (GPCR)–positive allosteric modulators (PAMs). 31 By measuring the initial velocity of fully activated c-Abl in the presence of 30 µM ATP in terms of substrate formation, we determined a normalized rate of 4.39 mP/min/nM ( Table 1 ). A grid titration experiment of ATP and c-Abl concentrations carried out for inactive c-Abl showed that <10% activation compared with fully activated c-Abl at all conditions tested ( Table 2 ). From this experiment, a final assay condition was chosen for HTS: 12.5 nM Abl, 10 µM ATP, 10-min activation time at ambient temperature, and 30-min IMAP reaction time ( Fig. 4 ).
The optimized IMAP assay demonstrated a window of ~80 mP and a robust Z′ above 0.6. DMSO tolerance experiments showed that although the mP window decreased slightly between 2.5% and 10% DMSO, the robust Z′ was maintained above 0.6 (data not shown). The reagent stability study showed that all reagents were stable at 4°C or RT for up to 7 h, and the assay signal was stable for up to 18 h (data not shown), which allowed ample time for screening.
An HTS campaign of 1.3 million compounds was performed under the above condition. In total, 8624 hits with greater than 30% response in the activation assay were identified in the primary screening. Of these, 5392 were confirmed by screening at 50 µM in duplicate, giving a 62% confirmation rate. This relatively low confirmation rate might be due to that fact that IMAP is an FP-based assay, and this format is subject to compound interference due to quenching, autofluorescence, or scattering. 32,33
Compared with other HTS campaigns of enzyme inhibitors or activators run within GlaxoSmithKline, the overall hit rate based on mean + 3 * SD was high (3.2% at a cutoff of 12% activation). This is probably due to 2 main factors. The first factor has to do with the high compound concentration in the activation step. In a typical enzyme inhibition screen, 10 µM of compound is used. However, in this screen, 50 µM of compound was incubated with c-Abl enzyme in the activation step to achieve 10 µM final at the point of c-Abl substrate addition. Screening at this concentration of compound would potentially yield nonspecific hits as discussed below. The second factor is likely the result of using the FP format for the HTS assay. As mentioned above, compound interference due to autofluorescence and scattering, especially at high concentration, tends to create false positives. It is essential that a counterscreen or an orthogonal assay be in place to triage the hits. Despite some interfering compounds, we successfully identified >1000 hits in the HTS. This outcome is better than other enzyme activator screens we ran previously (e.g., glucokinase activators; data not published). One unique feature of this screen was to allow a basal level (~10%) of phosphorylation-dependent activation of c-Abl. We believe this contributed to the success of the HTS.
As noted in the results section, 1024 compounds with pEC50 >5 and maximum activation of >50% were further subjected to analysis in a filter binding activation assay (see Materials and Methods) and a myristoyl peptide competition assay. 34 These experiments identified 579 compounds showing >2-fold activation in the filter binding assay. After submission to the myristoyl peptide competition assay, 15 showed >30% competition. In general, our data show that these actives fall into 2 major classes: (1) those that showed weak activity in the myristoyl peptide competition assay (referred to as type 1 hereafter) and (2) those that showed strong activity in the myristoyl peptide competition assay (referred to as type 2 hereafter).
As mentioned previously, under normal conditions, c-Abl is expressed in an autoinhibited conformation. Upon activation, the compact structure adopts an elongated shape where SH2 and SH3 domains dissociate from the kinase domain, allowing phosphorylation of Y245 and Y412. The proposed mechanisms for activating Abl include (1) displacement of N-terminal myristate, (2) displacement of the SH2 or SH3 domain, and (3) phosphorylation of Y245 in the SH2-kinase domain loop or Y412 in the activation loop. 19 The type 2 activators identified through the screen appear to act through direct displacement of N-terminal myristate. Upon displacement of the myristate, the c-Abl molecule undergoes a conformational change from a compact structure to an extended structure, which activates the kinase ( Fig. 4 ). It is worth noting that the recently published allosteric c-Abl inhibitor, GNFII, binds to the same myristoyl pocket. 35,36 Depending on the conformational state of C-terminal helix_I upon binding, compounds in this class can either inhibit or activate the kinase. 37 To elucidate the mechanism, we have crystallized our activators to c-Abl protein. 34 The mechanism of type 1 c-Abl activators is subject to future study.
The type 1 activators may work through mechanisms that do not involve direct displacement of the myristoyl binding. Although they may follow specific activation mechanisms that act through the binding of distinct sites other than the myristoyl pocket, we cannot exclude the possibility that some of these compounds may work through a promiscuous activation mechanism. Coan et al. 38 report that the mechanism of action of aggregate-based inhibitors is via partial protein unfolding when bound to an aggregate particle. Given that the test compounds were present at concentrations of 50 µM during the activation step, some compounds may have formed aggregates. The potential unfolding of the autoinhibited c-Abl conformation by the aggregates would therefore lead to enzyme activation. Another possibility is that if compounds show detergent-like property, they may affect c-Abl activity in the in vitro assay by altering the hydrophobicity of the reaction mixture.
To test whether these hits would show activity in whole cells, compounds of interest with IMAP pEC50 >6 ( Fig. 6A ) were evaluated using the ICW method. Many of the compounds active in the whole-cell assay had similar rank order potencies obtained in the biochemical assays, including the 5 series shown here ( Fig. 6B ). Interestingly, both type 1 and 2 hits showed active in the ICW assay (data not shown). Although some type 1 hits could be due to promiscuous mechanisms, it is likely that some hits may activate c-Abl kinase through a novel mechanism. Using this method, we demonstrated that it is possible to identify c-Abl kinase activators that do not bind to the myristoyl site.
In summary, we have developed a robust and sensitive 2-step IMAP assay to identify small-molecule activators of c-Abl kinase and used this assay to screen ~1.3 million compounds. More than 10 chemotypes were identified through the HTS, and these hits were shown to be active in both biochemical and cell-based assays. Further work on these series will involve structural modification(s) and further activity testing to boost compound potency prior to mechanism of action (MOA) analysis and proof-of-concept testing in preclinical animal models.
Footnotes
Acknowledgements
We thank Francesca Zappacosta in the Department of Analytic Biochemistry and Biophysics for phospho-mapping analysis, Sample Management Technology Group for their help with compound management, and Gordon McIntyre, Thomas Meek, and Allen Oliff for their support of carrying out this work.