
Abstract
The cardiac acetylcholine-activated K+ channel (IK,Ach) represents a novel target for drug therapy in the treatment of atrial fibrillation (AF). This channel is a member of the G-protein-coupled inward rectifier K+ (GIRK) channel superfamily and is composed of the GIRK1/4 (Kir3.1 and Kir3.4) subunits. The goal of this study was to develop a cell-based screening assay for identifying new blockers of the GIRK1/4 channel. The mouse atrial HL-1 cell line, expressing the GIRK1/4 channel, was plated in 96-well plate format, loaded with the fluorescent membrane potential-sensitive dye bis-(1,3-dibutylbarbituric acid) trimethine oxonol (DiBAC4(3)) and measured using a fluorescent imaging plate reader (FLIPR). Application of the muscarinic agonist carbachol to the cells caused a rapid, time-dependent decrease in the fluorescent signal, indicative of K+ efflux through the GIRK1/4 channel (carbachol vs. control solution, Z′ factor = 0.5-0.6). The GIRK1/4 channel fluorescent signal was blocked by BaCl2 and enhanced by increasing the driving force for K+ across the cell membrane. To test the utility of the assay for screening GIRK1/4 channel blockers, cells were treated with a small compound library of Na+ and K+ channel modulators. Analogues of amiloride and propafenone were identified as channel blockers at concentrations less than 1 µM. Thus, the GIRK1/4 channel assay may be used in the development of new and selective agents for treating AF.
Introduction
T
Cardiac arrhythmias are defined as abnormalities in the generation or conduction of electrical impulses in the heart. Atrial fibrillation (AF) is a rapid and irregular atrial muscle arrhythmia that results in erratic and incomplete cardiac contractions. AF is the most prevalent arrhythmia in the United States, affecting more than 2 million adult patients. 3,4 The frequency of AF increases with age, occurring in 3% to 5% of those older than 65 years of age and 9% of people older than 80 years. 3,4 Regardless of the underlying cardiac disease, chronic AF is associated with increased morbidity and mortality. Current antiarrhythmic drugs used in the treatment of AF are limited by suboptimal efficacy and a high incidence of toxicity. 3,4 One novel target for AF drug therapy is the GIRK1/4 channel. 4-6 Recent studies indicate that IK,Ach is constitutively active in patients with AF. 7 This constitutively active channel causes the atrial action potential duration to shorten with a resulting increase in cell excitability. 5,7 Therefore, GIRK1/4 channel blockers, by decreasing atrial excitability, should reduce the incidence of AF. However, discovery of new drugs that bind to and block the cardiac IK,Ach has been hampered by the absence of a cell-based screening assay that uses the GIRK1/4 channel.
In the present study, immortalized cardiac atrial cells, expressing the GIRK1/4 channel, were analyzed using patch-clamp recording procedures and fluorescent imaging plate reader (FLIPR) measurements. Application of carbachol activated a whole-cell IK,Ach current that was blocked by antiarrhythmic agents and the Kir toxin tertiapin. In cells loaded with the membrane potential–sensitive fluorescent dye bis-(1,3-dibutylbarbituric acid) trimethine oxonol (DiBAC4(3)), carbachol caused a rapid, time-dependent decrease in the fluorescent signal. This carbachol-induced decrease in fluorescence was blocked by BaCl2 and enhanced by increasing the driving force for K+ across the cell membrane. The GIRK1/4 channel/DiBAC4(3) assay was screened against a small-compound library of Na+ and K+ channel modulators. Analogues of propafenone and amiloride were identified as GIRK channel blockers at concentrations less than 1 µM. Thus, the GIRK1/4 channel assay will be useful for expanding the limited pharmacology of IK,Ach blockers and for developing new and selective agents for treating AF.
Materials and Methods
Cell culture and plating
The immortalized cardiac HL-1 cell line was generously supplied by Dr. William Claycomb (LSU Medical Center, New Orleans, LA). Cells were maintained in Claycomb/Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin, and L-glutamine (Sigma-Aldrich, St. Louis, MO). Myocytes were platted on gelatin-coated glass coverslips (5000 cells per coverslip; patch-clamp recording) and in black 96-well plates (25,000 cells per well; FLIPR measurements). Cells were stored in an incubator at 37°C (5% O2/95% CO2) and used on days 2 to 4 after plating.
Patch-clamp recording of IK,Ach currents
The patch-clamp method 8 was used to record the whole-cell IK,Ach using L/M EPC-7 (Adams & List Associates, Great Neck, NY) and Axopatch 200 (Axon Instruments, Sunnyvale, CA) amplifiers. Our procedures for measuring cardiac K+ channels have been described. 9 Pipettes were made from borosilicate glass capillaries (World Precision Instruments, Sarasota, FL) and had resistances of 1 to 2 Mohms when filled with internal solution. All experiments were conducted on isolated, noncoupled HL-1 cells at room temperature (22-24°C). For the measurement of IK,Ach, cells were placed in a normal Tyrode’s solution consisting of 132 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM dextrose, and 5 mM HEPES (pH 7.4; with NaOH) (280 mOsm). INa was blocked with tetrodotoxin (10 µM) and the Na+ channels inactivated by maintaining the myocytes at a holding potential of −40 mV. L-type Ca2+ channels were blocked with 500 nM nisoldipine. The internal solution consisted of 50 mM KCl, 60 mM K+-glutamate, 2 mM MgCl2, 1 mM CaCl2, 11 mM EGTA, 3 mM adenosine triphosphate (ATP), and 10 mM HEPES (pH 7.3; with KOH) (280 mOsm).
Membrane currents were recorded with 12-bit analog/digital converters (Axon Instruments). Data were sampled at 5 KHz and filtered at 1 KHz with a low-pass Bessel filter (Frequency Devices, Ottawa, IL). Series resistance was compensated to give the fastest possible capacity transient without producing oscillations. With this procedure, more than 70% of the series resistance could be compensated. Following the measurement of the HL-1 cell background current, IK,Ach was activated by the addition 10 µM carbachol using a rapid perfusion system. IK,Ach was defined as the BaCl2-sensitive current (see Fig. 1 ). IK,Ach blockers were added and inhibition determined immediately after carbachol-induced activation. This limited errors in quantifying drug block due to GIRK1/4 channel desensitization and rundown. Averaged current values presented are means ± SE.
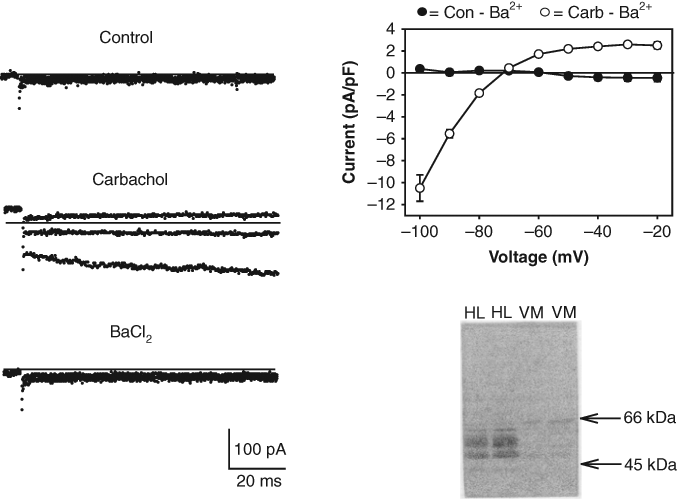
Expression of GIRK1/4 channels in HL-1 cells. Left panel: ion currents recorded during voltage steps applied from a holding potential of −40 mV to −100, −80 and −60 mV under control conditions, in the presence of 10 µM carbachol and following addition of 0.5 mM BaCl2. Solid lines represent zero current. Top right panel: I/V relationship for Ba2+-sensitive current activated by carbachol (Carb-Ba2+). Each point represents the mean ± SE of the current measured in 8 cells. Bottom right panel: immunoblot obtained with HL-1 cell (HL) and neonatal ventricular myocyte (VM) lysates using an anti-Kir3.1 Ab.
GIRK1 subunit Western blot analysis
Cell lysates for Western blot analysis were prepared by placing the HL-1 cell cultures into a lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 50 mM dithiothreitol, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mM phenylmethysulfonyl fluoride, 1 mM EGTA, 0.25% sodium deoxycholate, 2 µg/mL aprotinin, and protease inhibitor cocktail [Roche, Basel, Switzerland]). The protein content of the cell preparations was determined using a protein assay kit (Pierce, Rockford, IL). For Western blot analysis of the GIRK1 channel subunit, proteins were separated by electrophoresis on 8% sodium dodecyl sulfate (SDS) polyacrylamide gels using a mini PROTEAN cell (Bio-Rad, Hercules, CA). The running buffer contained 25 mM Tris, 193 mM glycine (pH 8.3), and 0.1% SDS. Proteins were transferred to polyvinylidene difluoride membranes using a Trans-Blot apparatus (Bio-Rad). The transfer buffer contained 25 mM Tris, 192 mM glycine (pH 8.5), and 20% methanol. For immunodetection, membranes were blocked in Tris-buffered saline (TBS) containing 0.1% Tween-20, bovine serum albumin, and 0.025% Na-azide for 60 min at room temperature. Antibodies to the Kir3.1 (GIRK1) channel (1:200) (Alomone, Jerusalem, Israel) were incubated with the membranes overnight at 4°C. After primary antibody treatment, the membranes were washed with TBS–0.1% Tween-20 and incubated with a secondary antibody (horseradish peroxidase [HRP]–conjugated rabbit IgG; Cell Signaling Technology, Danvers, MA). Immunoreactive bands were visualized on X-ray film (Kodak) using the enhanced chemiluminescence method (Pierce).
GIRK1/4 channel FLIPR assay
HL-1 cells were incubated for 1 h in Tyrode’s solution containing 5 µM DiBAC4(3) (AnaSpec, Fremint, CA) and fluorescent signals recorded using a Synergy2 microplate reader (BioTek, Winosski, VT) at 28°C. Test compounds were dissolved in DMSO at a stock concentration of 10 mM, diluted to various concentrations, and applied to the cells for 5 min prior to the fluorescent measurements. DiBAC4(3) was present in all experimental solutions. DMSO, up to 1% in Tyrode’s solution, had no adverse effects on the cell response. Carbachol or control solution (20 µL) was added to each well (total volume = 220 µL) at time zero using the Synergy2 injector system. Data points were collected at 10-s intervals over a 300-s sampling period at excitation and emission wavelengths of 480 and 520 nm, respectively. Positive (carbachol or carbachol + 1 mM BaCl2) and negative (water or DMSO) control experiments were performed to calculate Z′ factors. 10 The Z′ factor is defined as Z′ = 1 − (3σP + 3σN)/|µP − µN|, where µP and µN are the means of the positive control and negative control signals, and σP and σN are the standard deviations of the positive control and negative control signals, respectively. 10 Plates used for Z′ factor determination contained 2 rows each of positive and negative controls. Dose-response curves for selected compounds were obtained and drug potencies determined by fitting the data with the following curve: Max/(1 + ([drug]/IC50)k), where the IC50 is the concentration of the compound producing a 50% decrease in the maximal carbachol response (Max), and k is the slope factor. Interference of the test compounds with the DiBAC4(3) fluorescence was determined in the absence of cells by adding various concentrations of the compounds to Tyrode’s solution containing the dye.
Drugs and chemicals
Carbachol, KR-32568, antiarrhythmic agents, and an Na+, K+ channel modulator kit, containing 66 compounds, were purchased from Sigma-Aldrich. The propafenone analogues 1-{2-[3-(tert-butylamino)-2-hydroxypropoxy]phenyl}-3- phenyl-1-propanone, 1-{2-[2-(diethylamino)ethoxy]phenyl}-3- phenyl-1-propanone, 1-{2-[3-(diethylamino)-2-hydroxypropoxy]phenyl}-3-phenyl-1-propanone, 5-hydroxy propafenone, 1-{2- [2-hydroxy-3-(1-piperidinyl)propoxy]phenyl}-3-phenyl-1- propanone, 1-{2-[2-hydroxy-3-(4-morpholinyl)propoxy]phenyl}- 3-phenyl-1-propanone, 1-{2-[3-(ethylamino)-2-hydroxypropoxy]phenyl}ethanone, and 1-(1-piperidinyl)-3-(4-propylphenoxy)-2-propanol were obtained from ChemBridge Corp. (San Diego, CA).
Results
Measurement of IK,Ach currents in HL-1 cells
IK,Ach is highly expressed in cardiac sinoatrial nodal, atrioventricular nodal, and atrial tissues. 1,2 HL-1 cells are an immortalized atrial cell line that displays an adult-like cardiac genotype and contracts spontaneously in cell culture. 11,12 Figure 1 displays IK,Ach currents measured in the HL-1 cells using the whole-cell arrangement of the patch-clamp technique. Cells were bathed in normal Tyrode’s solution (5 mM K+), and the pipette contained a KCl/K+-glutamate (140 mM K+) solution. Application of carbachol to the recording chamber resulted in the activation of an inward rectifying current ( Fig. 1 ). As expected for an inward rectifier K+ channel, the carbachol-activated current was completely blocked by BaCl2 ( Fig. 1 ). The reversal potential (Erev) for the current (−72 mV) was within range of the calculated Nernst equilibrium potential (EK) (−85 mV) for a K+-selective channel under these conditions.
The cardiac IK,Ach is composed of the inward rectifier Kir3.1 and Kir3.4 (GIRK1 and GIRK4) subunits. 1,2 As shown in Figure 1 , an immunoreactive doublet protein corresponding to the approximately 50- to 55-kDa GIRK1 subunit was identified in HL-1 cells but not in ventricular myocytes using an anti-GIRK1 antibody. The pharmacological properties of the HL-1 cell IK,Ach current were also examined. A number of agents, including the Kir toxin tertiapin (100 nM) and the antiarrhythmic drugs flecainide (100 µM) and propafenone (10 µM), block IK,Ach in primary cultures of adult atrial myocytes. 13-15 All 3 of these agents strongly blocked IK,Ach at the concentrations tested ( Fig. 2 ). In contrast, addition of control solution (containing drug vehicle) produced no significant change in the current ( Fig. 2 ). The full dose-response curve for the propafenone block of IK,Ach is included in Figure 2 . The IC50 for the propafenone block was 2 µM. Thus, the inward rectifying current versus voltage relationship, the Ba2+-sensitivity and pharmacology of the whole-cell IK,Ach current, and the presence of Kir3.1 subunits are all consistent with the expression of GIRK1/4 channels in the HL-1 cells.
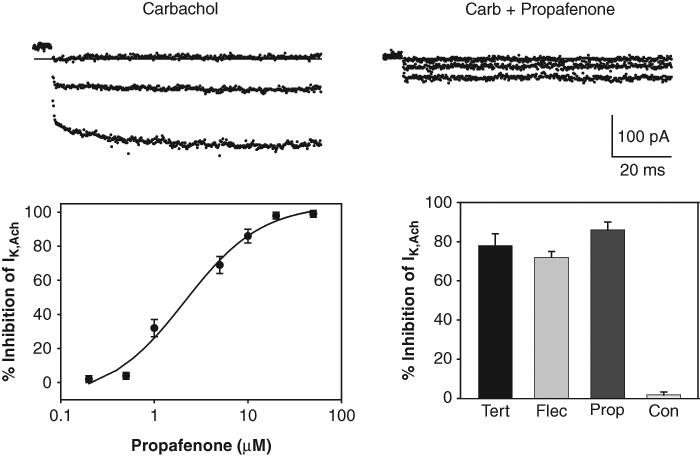
Pharmacological properties of the HL-1 cell IK,Ach. Top panels: IK,Ach recorded during voltage steps applied to −100, −80, and −60 mV in the presence of carbachol and following the addition of 10 µM propafenone. Bottom left panel: dose-response curve for block of IK,Ach measured at −100 mV with propafenone (IC50 = 2 µM). Each point represents the mean ± SE inhibition measured in 4 to 6 HL-1 cells. Bottom right panel: percent inhibition of IK,Ach measured at −100 mV with tertiapin (Tert) (100 nM), flecainide (Flec) (100 µM), propafenone (Prop) (10 µM), and control solution. Each bar represents the mean ± SE inhibition measured in 6 HL-1 cells.
Development of the GIRK1/4 channel FLIPR assay
Having demonstrated that the IK,Ach current–GIRK1/4 channel is expressed in the HL-1 cell line, it was determined if these cells could be used for FLIPR measurements of the channel. The experimental design of the screening assay is outlined in Figure 3 . HL-1 cells were cultured in 96-well plates and loaded with the fluorescent membrane potential–sensitive dye DiBAC4(3). Membrane potential–sensitive dyes such as DiBAC4(3) distribute across the plasma membrane when cells are in the rested state and reach equilibrium ( Fig. 3 , top panel). The DiBAC4(3) molecules inside the cells become strongly fluorescent upon binding to intracellular proteins and other cytoplasmic components. Treatment of the cells with carbachol activates Gi, stimulating the GIRK1/4 channels to open (bottom panel). The resulting efflux of K+ out of the cell should therefore cause the resting membrane potential to become more negative and the DiBAC4(3) molecules to redistribute to the outside of the cell. As a result, the cell fluorescent signal should decrease.
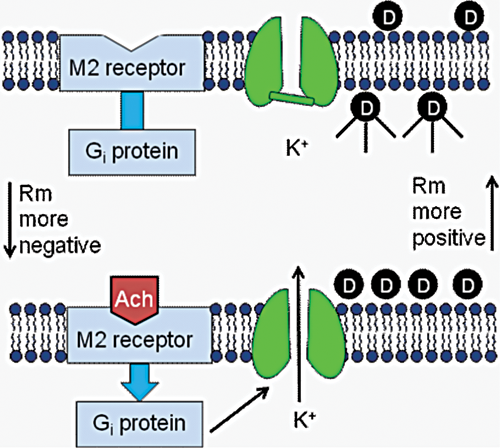
Development of a screening assay for identifying new GIRK1/4 channel blockers. See text for details.
The applicability of the GIRK1/4 channel fluorescent assay for drug testing is evaluated in Figure 4 . Each figure plots the DiBAC4(3) fluorescence signal measured over time in the 96-well plates using a Synergy2 microplate reader (BioTek). As predicted by the experimental model, addition of carbachol (10 µM) to the cardiac cells caused a rapid, time-dependent decrease in the DiBAC4(3) fluorescent signal ( Fig. 4 , top left panel). In contrast, addition of control solution produced only a small instantaneous rise in the fluorescence ( Fig. 4 ). Qualitatively similar results were obtained using the oxonol dye HLB 021-152 (AnaSpec; results not shown). Comparison of peak fluorescent changes in wells injected with control and carbachol solutions gave Z′ factors ranging from 0.50 to 0.64 (4 plates from 2 experimental days). The peak carbachol-induced fluorescent signal obtained from these plates was 0.956 ± 0.008 (mean ± SD). To quantify the GIRK1/4 channel fluorescent signal, we subtracted the averaged control measurement, obtained in each 96-well plate, from the records measured in the presence of carbachol (Carb – Con; Fig. 4 ). This control-subtracted fluorescent signal (i.e., the carbachol- sensitive component) was subsequently obtained and analyzed throughout the rest of the study.
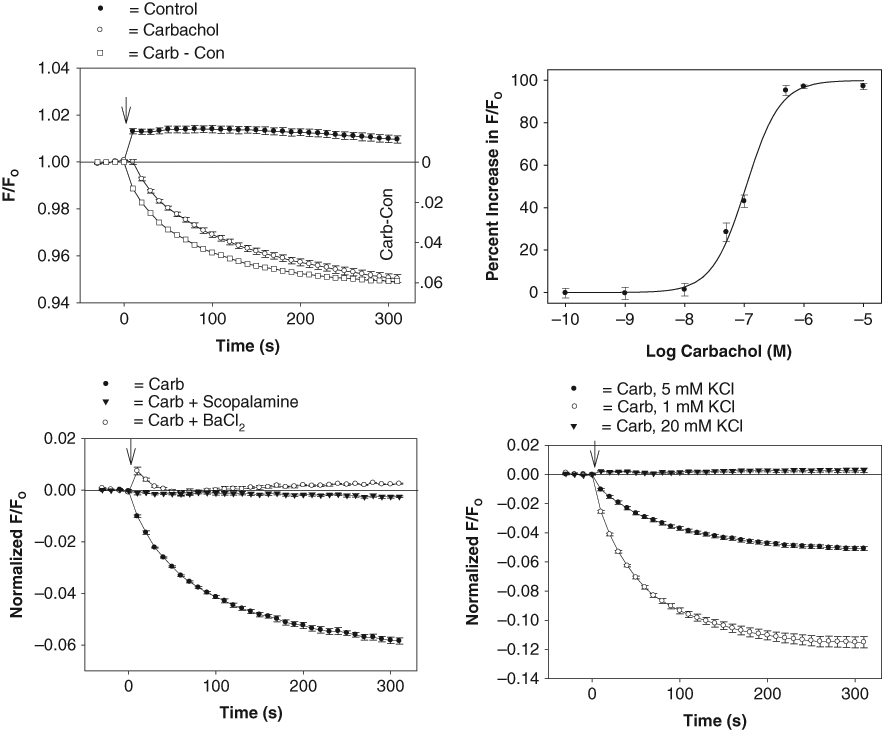
Development of a GIRK1/4 channel blocker screening assay. Top left panel: DiBAC4(3) fluorescent intensities obtained in HL-1 cells in the presence or absence of carbachol. The ratio of the fluorescent intensity (F/FO) was calculated by dividing the signal in the presence (F) of carbachol (or control solution) by the baseline signal measured before (FO) addition of carbachol (or control solution). Each point represents the mean ± SE obtained in 6 wells. Carbachol was added at time zero (↓). Top right panel: dose-response curve for the carbachol-sensitive fluorescent signal (EC50 = 110 nM). Each point represents the mean ± SE of 10 measurements. Bottom left panel: carbachol fluorescent signal measured in HL-1 cells pretreated for 5 min with vehicle solution (H2O) (Carb) and either 1 µM scopolamine or 1 mM BaCl2. Each point represents the mean ± SE obtained in 6 to 8 wells. Bottom right panel: carbachol fluorescent signal measured in HL-1 cells in Tyrode’s solutions containing 1 mM, 5 mM, and 20 mM KCl. Each point represents the mean ± SE obtained in 6 wells.
The pharmacological and biophysical properties of the carbachol-sensitive fluorescent signal are summarized in Figure 4 . Carbachol activated the GIRK1/4 channel in a concentration-dependent manner with a half-maximal effective concentration (EC50) of 110 nM ( Fig. 4 , top right panel). The carbachol signal was blocked by 1 mM BaCl2 and inhibited during pretreatment with the muscarinic antagonists scopolamine (1 µM; Fig. 4 , bottom left panel) and atropine (1 µM; results not shown). The effect of altering the driving force for K+ across the plasma membrane on the GIRK1/4 channel signal was also determined with the HL-1 cells. Carbachol-induced changes in the DiBAC4(3) signal were measured in Tyrode’s solutions containing 1, 5, and 20 mM KCl. Under these conditions, the reversal potential (Erev) for the GIRK1/4 channel should follow EK and approach −120 mV and −50 mV in 1-mM and 20-mM KCl solutions, respectively. As shown in Figure 4 (bottom right panel), reducing the extracellular K+ concentration ([K]o) from 5 mM to 1 mM produced a 2-fold increase in the carbachol-sensitive signal (mean = 2.0 ± 0.1, 3 plates). In contrast, increasing [K]o to 20 mM, which should shift EK close to the resting potential of the HL-1 cell (−50 to −65 mV), 16 completely eliminated the carbachol-mediated decrease in the fluorescent signal.
Identification of GIRK1/4 channel blockers
IK,Ach is blocked by several antiarrhythmic drugs, including propafenone, flecainide, and amiodarone. 13-15 Therefore, the GIRK1/4 channel assay was screened against a group of antiarrhythmic agents. Propafenone was the most potent of the drugs tested, with an IC50 of 3 µM ( Fig. 5 ). This inhibitory potency was similar to that measured for the propafenone block of the IK,Ach current (IC50 = 2 µM; Fig. 2 ). Several structural analogues of propafenone were obtained from ChemBridge (see Materials and Methods) and tested for their inhibitory action. Of these compounds, only 1-{2-[3-(tert-butylamino)-2-hydroxypropoxy] phenyl}-3-phenyl-1-propanone (TBPP; IC50 = 2 µM) was more potent than propafenone ( Fig. 5 ). Other antiarrhythmic agents, including ibutilide (IC50 = 16 µM), quinidine (IC50 = 24 µM), and flecainide (IC50 = 56 µM), were also effective in inhibiting the carbachol-sensitive fluorescence signal. In contrast, drugs such as sotalol (blocker of the rapid component of the cardiac-delayed rectifier K+ channel), glybenclamide (ATP-sensitive K+ channel blocker), and procainamide (voltage-gated Na+ channel blocker) caused no block at the concentrations tested ( Fig. 5 ). Amiodarone, another antiarrhythmic agent that blocks K+ channels, 14 was not evaluated since it interfered with the DiBAC4(3) fluorescence at concentrations equal to or greater than 1 µM.
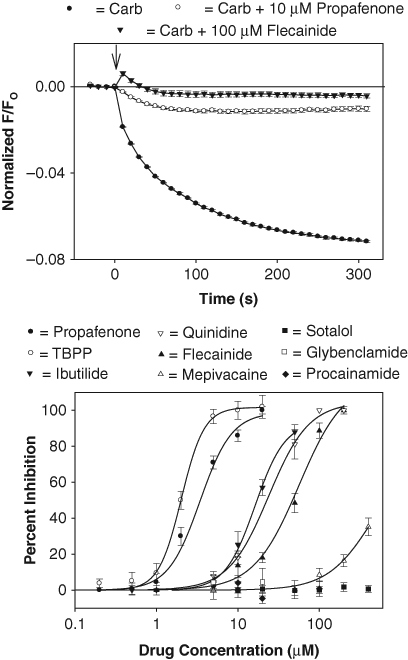
Screen of antiarrhythmic agents using the GIRK1/4 channel blocker assay. Top panel: carbachol-sensitive fluorescent signal measured in cells pretreated for 5 min with drug vehicle solution (DMSO) (Carb) and either 10 µM propafenone or 100 µM flecainide. Each point represents the mean ± SE value obtained in 6 wells in 1 plate. Bottom panel: dose versus response curve for drug block of the carbachol signal. Each point represents the mean ± SE inhibition obtained from 3 to 5 experiments such as the one displayed in the top panel. Calculated IC50 values were 1-{2-[3-(tert-butylamino)-2-hydroxypropoxy] phenyl}-3-phenyl-1-propanone (TBPP) = 2 µM, propafenone = 3 µM, ibutilide = 16 µM, quinidine = 24 µM, and flecainide = 56 µM. Glybenclamide was not tested at concentrations >20 µM due to interference with the DiBAC4(3) fluorescence.
A small-compound library, consisting of Na+ and K+ channel modulators (Sigma-Aldrich), was also tested using the GIRK1/4 channel assay. Several analogues of the epithelial Na+ channel blocker amiloride were identified as inhibitors of the carbachol-sensitive signal when the modulators were screened at 1 µM. The full dose-response curves for these drugs are plotted in Figure 6 . Both 5-(N-ethyl-N-isopropyl)-amiloride (EIPA; IC50 = 0.8 µM) and 5-(N, N-hexamethylene)-amiloride (HMA; IC50 = 0.9 µM) were effective at concentrations below 1 µM. In contrast, amiloride caused only a partial block at concentrations up to 100 µM ( Fig. 6 ). Since EIPA and HMA inhibit the Na+/H+ exchanger (NHE), 17 the effect of the selective NHE inhibitor KR-32568 ([5-(2-methyl-5-fluorophenyl)furan-2-ylcarbonyl] guanidine) was determined in the assay. However, at drug concentrations up to 1 µM, KR-32568 caused no significant change in the carbachol-sensitive fluorescence ( Fig. 6 ).
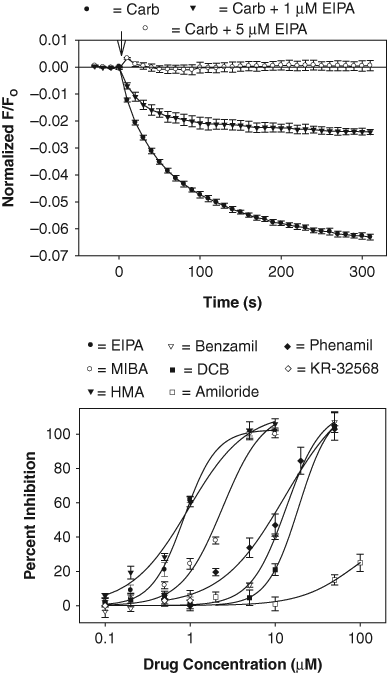
Screen of amiloride analogues using the GIRK1/4 channel blocker assay. Top panel: carbachol-sensitive fluorescent signal measured in cells pretreated for 5 min with drug vehicle solution (DMSO) (Carb) and either 1 µM or 5 µM 5-(N-ethyl-N-isopropyl)-amiloride (EIPA). Each point represents the mean ± SE value obtained in 6 wells in 1 plate. Bottom panel: dose versus response curve for drug block of the carbachol signal. Each point represents the mean ± SE inhibition obtained from 3 to 5 experiments such as the one displayed in the top panel. Calculated IC50 values were EIPA = 0.8 µM, 5-(N, N-hexamethylene)-amiloride (HMA) = 0.9 µM, 5-(N-methyl-N-isopropyl)-amiloride (MIBA) = 2 µM, benzamil = 13 µM, phenamil = 13 µM, and dichlorobenzamil (DCB) = 20 µM. KR-32568 was not tested at concentrations >1 µM due to interference with the DiBAC4(3) fluorescence.
Discussion
Expression of GIRK1/4 channels in HL-1 cells
The present study demonstrates that immortalized cardiac HL-1 cells can be used for identifying new GIRK1/4 channel blockers. Consistent with the presence of the GIRK1/4 channel, HL-1 cells displayed a carbachol-activated Kir current and expressed the Kir3.1 channel subunit. Application of carbachol to cells loaded with a membrane potential–sensitive dye caused a rapid, time-dependent decrease in the fluorescent signal that was inhibited by BaCl2 and muscarinic receptor antagonists. Finally, structural analogues of propafenone and amiloride were identified as GIRK1/4 channel blockers at concentrations less than 1 µM.
The traditional approach to ion channel drug discovery has involved the expression of recombinant channels in heterologous cell lines such HEK-293 and Chinese hamster ovary (CHO) cells. However, these cells often lack the relevant “physiological environment” that is present in native tissues and required for normal pharmacological responses. 18,19 For example, the expression profile of G-proteins and ion channels in HEK-293 cells differs from that found in muscle and nerve. 18,19 Thus, recent studies have examined the suitability of primary cells, as well as clonal cell lines derived from animal tissues, for drug discovery. 19 AT-1 atrial myocytes were the first transformed cardiac cell line to be developed. 20 AT-1 cells were derived from a transgenic mouse that expresses the atrial natriuretic factor–inducible SV40 large T antigen. 20 HL-1 and HL-5 cells were subsequently derived from the AT-1 cells as cardiac cell lines that could be serial passaged after culture. 11,12 These cells display an adult-like cardiac genotype and contract spontaneously in cell culture. 11,12 In addition, cardiac G-protein-coupled receptors (GPCRs), cell signaling molecules, and voltage-gated ion channels are present in the cells. 11,12 Since the human and mouse M2 receptors and Kir3.4 subunits share more than 90% homology, 1,2 HL-1 cells represent an ideal preparation for cardiac drug discovery.
IK,Ach is highly expressed in cardiac sinoatrial nodal, atrioventricular nodal, and atrial tissues. 1,2 In contrast, IK,Ach is expressed at a much lower density in cardiac ventricular tissue. 2 IK,Ach currents have been extensively characterized in primary cultures of atrial myocytes obtained from humans, canines, rabbits, and rodents. 2 Two previous studies have provided supportive preliminary data for the expression of GIRK1/4 channels in the HL-1 and HL-5 cell lines. Xiao et al. 16 demonstrated that HL-5 cells express M2 receptors and a muscarinic-activated K+ current. Stimulation of the M2 receptor with carbachol reduced both the amplitude and duration of the HL-5 cell action potential. 16 Baki et al. 21 reported in abstract form that HL-1 cells express Kir3.x subunits and display electrophysiological properties consistent with the atrial IK,Ach. However, neither of these studies provided any pharmacological data on the GIRK1/4 channel, nor was the suitability of the cells for drug discovery examined.
Development of a GIRK1/4 channel blocker assay
The goal of the present study was to develop a screening assay for identifying new GIRK1/4 channel blockers. For this purpose, HL-1 cells were cultured in 96-well plates, loaded with the fluorescent membrane potential–sensitive indicator DiBAC4(3), and stimulated with carbachol. Membrane potential–sensitive dyes have been used in drug discovery for a wide variety of ion channels, including K+, 22,23 Na+, 24,25 hyperpolarization-activated cyclic-nucleotide gated, 26 and transient receptor potential 27 channels. Although fluorescent oxonol dyes, such as DiBAC4(3), provide a convenient and inexpensive approach for measuring changes in membrane potential, they are limited by their slow response compared with electrophysiological measurements. In addition, oxonol dyes can be sensitive to changes in temperature and can be perturbed by test compounds. In the present study, several drugs, including glybenclamide (>20 µM), KR-32568 (>2 µM), and amiodarone (≥1 µM), reduced the DiBAC4(3) fluorescence when tested under cell-free conditions. Wolff et al. 28 evaluated the use of several membrane potential probes, including oxonol dyes and fluorescence resonance energy transfer (FRET) dye systems, for measuring drug block of an endogenous Kir channel in RBL-2H3 cells. Although some compounds did produce oxonol dye interference when tested at concentrations ≥10 µM, comparative IC50 values for 8 different Kir channel blockers were obtained using the different membrane potential probes. Nonetheless, a thorough evaluation of other membrane potential–sensitive dyes will be necessary before the HL-1 cell GIRK1/4 channel assay can be converted to a high-throughput system. Alternate procedures, such as the thallium influx assay, have been used to identify GPCRs linked to GIRK channels. 29
Use of GIRK1/4 blockers in atrial fibrillation
AF is the most commonly occurring cardiac arrhythmia and is responsible for significant morbidity, mortality, and health care costs. Several recent studies have identified IK,Ach as a novel therapeutic target for AF. 4-6 However, the development of new and specific blockers of IK,Ach has been limited due to the absence of a cell-based screening assay for the GIRK1/4 channel. A number of antiarrhythmic agents, including amiodarone, flecainide, and propafenone, block IK,Ach in primary cultures of adult atrial myocytes. 13-15 Consistent with this, we found that flecainide and propafenone blocked both the HL-1 cell IK,Ach and carbachol-sensitive fluorescent signal. For propafenone, there was a good correlation between drug potency measured with the patch-clamp procedure (IC50 = 2 µM) and the FLIPR (IC50 = 3 µM). The IC50 values determined in the fluorescent assay for propafenone and flecainide (IC50 = 56 µM) are consistent with the IC50 values measured in primary cultures of guinea pig atrial myocytes (1 and 20 µM, respectively) using patch-clamp analysis. 13 As shown in Figure 6 , 3 structural analogues of amiloride inhibited the carbachol-sensitive fluorescent signal at concentrations less than 1 µM. Acyl guanidine compounds such as EIPA, 5-(N-methyl-N-isopropyl)-amiloride (MIBA), and HMA inhibit the Na+/H+ exchanger (NHE). 17 Thus, these and chemically related drugs (cariporide, eniporide, etc.) may provide a good starting point for structure versus potency studies. Recently, 2 benzopyran derivatives, NIP-142 and NIP-151, were demonstrated to block GIRK channels expressed in HEK-293 cells at nanomolar concentrations. 30,31 When tested in a canine model of AF, NIP-151 decreased atrial excitability and converted AF to sinus rhythm. 30,31 Although the selectivity of these agents for the GIRK family of IKr channels was not established, the results of these studies support the further development of new and selective GIRK1/4 channel blockers for the treatment of AF.
Footnotes
Acknowledgements
The author thanks Ms. Kathy Long, Ms. Jining Zhang, and Mr. Zachary Coffman for their excellent technical assistance. This work was supported by US Public Health Service award HL-45789 and a grant from the University of South Carolina.