
Abstract
High-content screening (HCS) technologies are becoming increasingly used in both large-scale drug discovery and basic research programs. These automated imaging and analysis technologies enable the researcher to elucidate the complex biology that underlies the functions of genes, proteins, and other biomolecules at the cellular level. HCS combines the power of automated digital microscopy and advanced software-based image analysis algorithms to detect and quantify biological changes in cells and tissues. This technology is a particularly powerful tool when used to interrogate the cellular effects of exogenously applied agents such as RNAi and/or small molecules. HCS allows for the evaluation of cellular perturbations that occur both at the level of the single cell and within cellular populations. In a multivariate approach, multiple cellular parameters are collected, allowing for more complex analysis. However, in these scenarios, data flow and management still represent substantial bottlenecks in HCS projects. HCS data include a diversity of information from multiple sources such as details pertaining to screening libraries (e.g., siRNA and small molecules), image stacks acquired from automated microscopes (of which there may be up to several million), and the image analysis data. From this, postprocessing algorithms are required to generate statistical, quality control bioinformatic information and ultimately a final hit list. To accomplish these individual tasks, numerous tools can be used to perform each analytical step; however, management of the entire information flow currently requires the use of commercially available proprietary software, the scope of which is often limited, or bespoke customized scripts. In this article, the authors introduce an open-source research tool that allows for the management of the entire data flow of the HCS data chain, by handling and linking information and providing many powerful postprocessing and visualization tools.
Keywords
Introduction
O
Despite the power of the high-content approach, more conventional screening strategies do not offer the sensitivity to identify all hits, particularly in screens with a complex readout. For example, using genome-scale libraries, which typically contain 2 to 4 small interfering RNAs (siRNAs) targeting each gene, it is difficult to systematically identify genes for which multiple siRNAs are active across a screen—that is, the effects of which do not fall within an upper threshold of a given response (moderately active siRNAs). In such cases, it is expected that the biological implications of disrupting an individual gene function using siRNA will result in a common phenotypic response or pattern of responses across the screen. To investigate such patterns in a sensitive manner, a good data-mining package is required. 3
Many free and commercial software packages are now available to analyze and manage HCS data sets, although it is still difficult to find a single off-the-shelf software package that covers all aspects of the HCS workflow. Pipeline (workflow) systems are now becoming a crucial requirement for enabling biologists to perform large-scale HCS experiments with very large data sets. Currently, there are a few suitable workflow systems available for such applications; examples of these are Kepler, 4 Taverna, 5 InforSense KDE, 6 and Pipeline Pilot. 7
Taverna has been developed to integrate Web services by workflows, and this is specified in a choreography language: XML Simple Conceptual Unified Flow Language. With Taverna, the editor is embedded within its engine in a Java stand-alone application. One drawback of Taverna is that it is a “heavy” package and, as such, difficult for an end user to download.
Pipeline Pilot was one of the very first workflow systems used in the life sciences arena. This system is chemically intelligent and possesses a robust and highly scalable environment that can run on large-scale Linux clusters. Pipeline Pilot is widely used to process drug discovery data and comes with specialized solutions for computational chemistry, chemoinformatics, and bioinformatics. InforSense KDE environment and its open workflow technology provides an excellent workflow system with specialized extensions such as BioSense, ChemSense, and TextSense. BioSense covers high-performance bioinformatics solutions ranging from sequence analysis to microarray informatics and remote database annotation. ChemSense provides a large range of chemoinformatics solutions ranging from the analysis and visualization of chemical libraries to the development of combinatorial chemistry libraries and includes a wide range of QSAR, ADME-Tox prediction, molecular modeling, and evaluation methods. Kepler is another workflow-based system and has been used in various scientific domains, including molecular biology. The current version of Kepler provides full support for computational chemistry and statistical analysis. Core Kepler tool has General Atomic and Molecular Electronic Structure System (GAMESS) as an ab initio quantum chemistry package. Kepler’s ability to interface with programs that need command line invocation has made it an excellent choice for computational chemistry calculation workflows.
However, due to the high costs involved with the above workflow systems or, in some cases, a lack of full HCS plug-ins integration, these packages are still not accessible to many research institutions working on HCS experiments. In this article, we present the HCDC-HITS package, a visual programming environment for HCS data mining, data management, and an execution engine for image-processing composition (
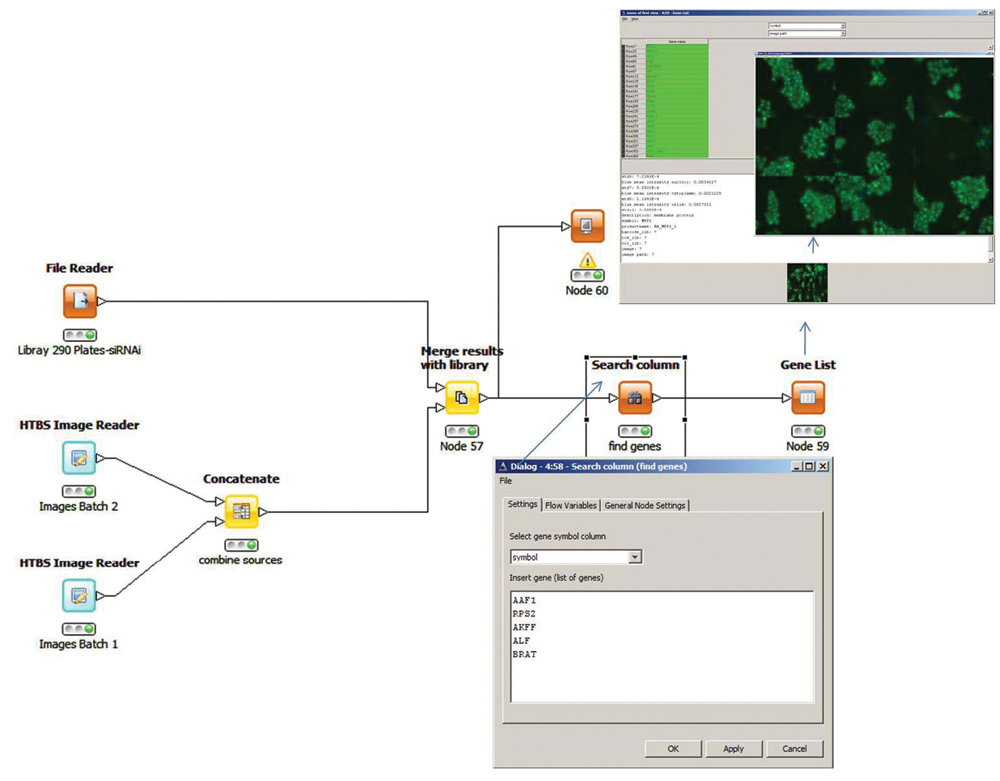
HCDC-HITS workflow example. RNAi library information is concatenated with image data and image-processing parameters. The search node allows 4 oligos for a specific gene to be found together with all related data from screening experiments. Image data are visualized with an integrated channel filter.
Workflow System for HCS
The concept of workflow is not new, having been used by many organizations, over several years, to improve productivity and increase efficiency. A workflow system is highly flexible and can accommodate any changes or updates as and when new or modified data and corresponding analytical tools become available. A workflow environment allows biologists to perform the integration themselves without involving any programming.
Workflow systems are different from programming scripts and macros in one important respect. Programming systems and macros use text-based languages to create lines of code, whereas applications such as HCDC-HITS use a graphical programming language. The workflow in HCDC-HITS is termed abstract in that it is not yet fully functional, but the actual components are in place and in the requisite order. In general, workflow systems concentrate on creation of abstract process workflows to which data can be applied when the design process is complete. In contrast, workflow systems in the life sciences domain are often based on a data flow model due to the data-centric and data-driven nature of many scientific analyses. A comprehensive understanding of biological phenomena can be achieved only through the integration of all available biological information and different data analysis tools and applications. A workflow environment allows HCS researchers to perform the integration themselves without involving any programming. As such, the workflow system allows the construction of complex in silico experiments in the form of workflows and data pipelines. Data pipelining is a relatively simple concept. Any computational component or node has data inputs and data outputs. Data pipelining views these nodes as being connected together by “pipes” through which data flow (
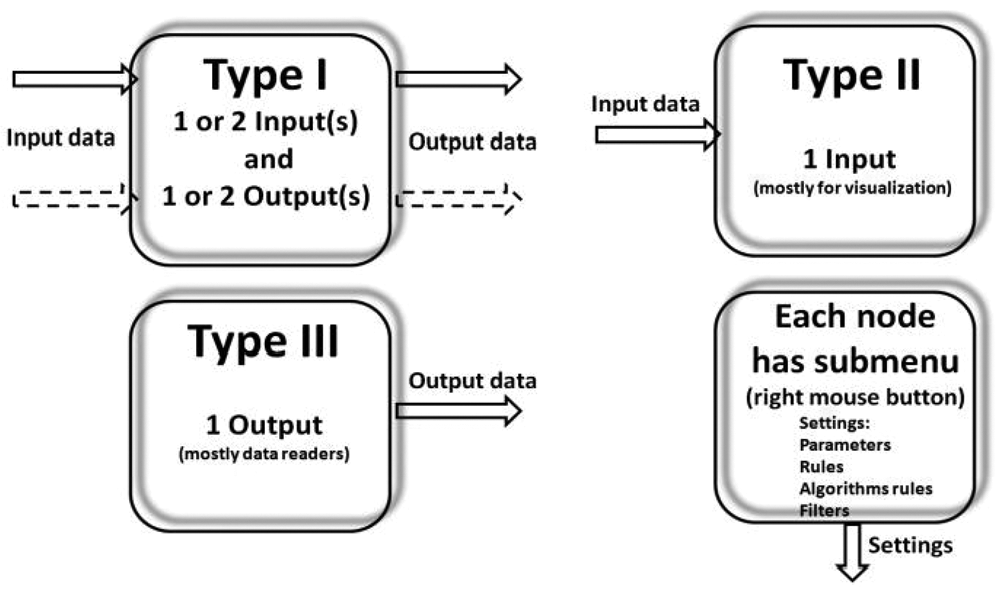
General concept of a pipeline node. The component properties are described by the input metadata, output metadata, and user-defined parameters or transformation rules. The input and output ports can have 1 or more incoming or outgoing metadata or images.
HCDC-HITS builds a flow by dragging and dropping nodes from the Node Repository into the main panel and connecting them. Nodes are the basic processing units of a workflow (
In a workflow-controlled data pipeline, as data flow, they become transformed (i.e., raw data are analyzed to become information, and the collected information gives rise to knowledge). A workflow system is highly flexible and is designed to accommodate any changes or updates that are required when new or modified data and corresponding analytical tools become available.
Data analysis in HCDC-HITS
In the workflow optimization phase illustrated in
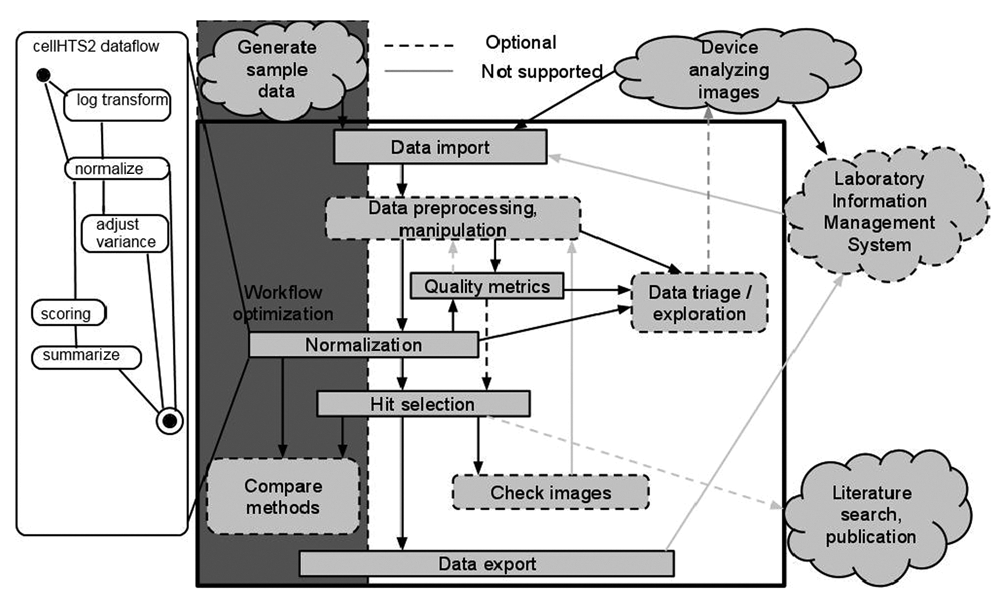
Overview of the data analysis process in HCDC-HITS.
The first step in the data analysis process involves the import of data using the File Reader node, which may be followed by some preprocessing/manipulation of the raw data (e.g., by combining different parameters subtracting background, computing ratio of channel measures, etc.). Following this, quality metrics may be performed, and these may be generated from the normalized values. The process labeled “normalization” may be split into multiple steps—for example, log transformation, normalization, variance adjustment, scoring, and summarization (see below). The optimum method of normalization may be chosen during the optimization phase. Following normalization of the data, hit selection may commence, and multiple strategies are supported for this. Finally, following hit selection, the user may revert to the original images to correlate with generated measurements and to check for any anomalies.
HCDC-HITS and cellHTS2 integration
CellHTS2 9 is a tool that is suitable for analysis of cell-based screens as it supports different methods of normalization, as well as scoring and summarization methods, all of which are used frequently in high-content screens. Prior to the analysis of multiparametric screens with different classification methods, data must be normalized to achieve a comparable range of values. Without this step, some of the parameters would dominate the analysis, or phenomena such as edge effects may skew the data.
Most HCS statistical methods 10 are available in HCDC-HITS. In addition to this, every method supported by cellHTS2 is also available in HCDC-HITS. Examples of these are multiplicative or additive median of samples/negative controls, multiplicative or additive mean of samples, multiplicative percent of (positive) controls, normalized percent inhibition, B score, and Loess/locfit. Each normalization method mentioned can be preceded by a log2 transformation. The variance adjustment of the screen plates is performed either on physical plates or within each replicate line based on the median absolute deviation of normalized sample values. Scoring of values is performed on each replicate line or on physical plates, and the following options are available: no scoring, robust z-score (both replicate lines and physical plates), nonrobust z-score (both replicate lines and physical plates), and normalized percent inhibition (only on replicate groups). Summarization of scored values allows the user to compute the scored replicate values, one single value for each well. The following options are available: mean of the scores, median of the scores, minimal score, maximal score, root mean square of the scores, closest score to zero, and farthest score from zero. It should also be pointed out that linking these data sets to results, based on the already available information, is possible using the R-based biomaRt 3 package.
To allow for an automatable evaluation of a large combination of both feature selection and classification methods, we have used the HCDC-HITS package and integrated the Weka library. HCDC-HITS is implemented in Java, and its architecture strictly follows a workflow-based operational model. We have developed a set of HCS/HTS and Library Handling nodes for HCDC-HITS that provide the tools necessary for all HCS pattern recognition tasks. Available HCS nodes include a filtering function for data preprocessing, a feature selection node offering filter methods, and training nodes to build classification models using training data. To allow for a systematic benchmark of different pattern recognition methods, we implemented a so-called training node. It is possible to save classifiers, which have been built using the training node, for use with new data sets. All nodes provide a graphical user interface. A screenshot of the batch data processing using HCDC-HITS nodes is shown in
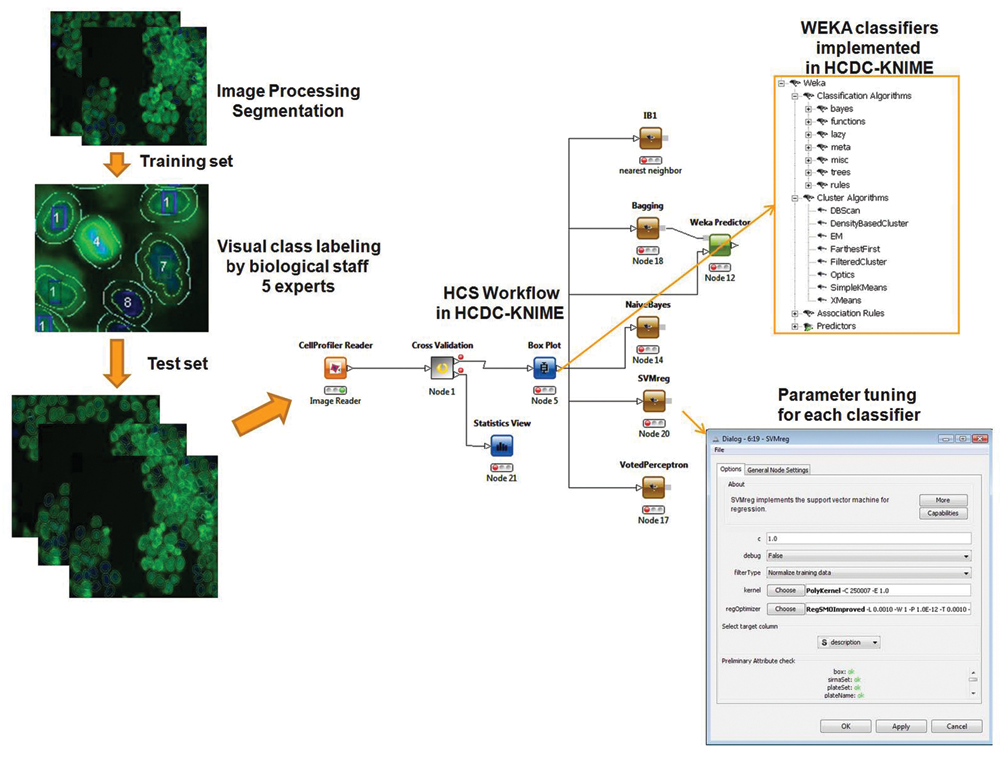
Analysis schema and screenshot of the HCDC-HITS workflow including Weka nodes for high-content screening pattern recognition.
For our evaluation of different classification methods, it was possible to use different classifiers from the HCDC-HITS Weka nodes. We used a k-nearest neighbor (kNN) classifier, a one-rule classifier, a naive Bayes classifier, a C4.5 decision tree, SVM (radial base function, RBF kernel), and voted perceptron. All classifiers were trained using the HCDC-HITS node default configuration parameters (
HCS Components in HCDC-HITS
HCDC-HITS provides innovative automated processes that require minimal manual intervention. The algorithm uses predefined modules called nodes for individual tasks such as data import, processing, or visualization. This system will also permit the selection of any number of nodes and the design of the data flow between these elements (
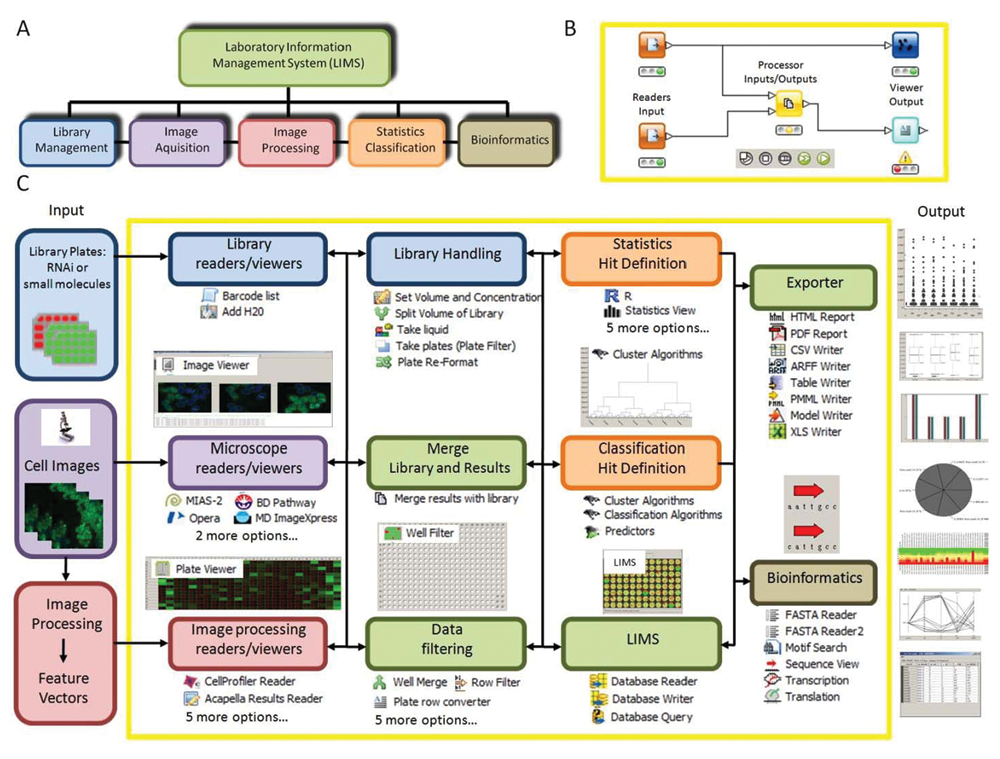
HCDC-HITS platform. (
Library handling, library readers
These components allow for the registration of dilution and volume changes during liquid handling and the management of barcode information. Library information presented in many formats can be used to identify a sample within a library of RNAis or small compounds.
Microscope and image-processing readers and viewers
HCDC-HITS can import microscopy images in all popular formats and retrieve data generated by image-processing software. These include the Acapella and Cellenger BioApplications, as well as open-source programs such as CellProfiler. 12
Visualization and export tools
Imported and processed data can be visualized by image or data browsers at each stage of the processing pipeline and exported in many formats.
Laboratory Information Management System (LIMS)
Efficient LIMS are available and are integrated into HCDC-HITS; this enables management of HCS information, which can be saved to a database.
Data filtering and processing, statistics and classification
These nodes allow the user to define the hits of a screen as an ultimate output. HCDC-HITS supports data processing in many ways as well as filtering or thresholding and can also employ machine-learning approaches.
Data integration
We developed nodes for the seamless integration of library data with image information, numerical results, and metadata across experiments.
Quality control
Because direct supervision of experiments is not feasible in HCS, HCDC-HITS offers modules that deal with assay robustness and quality control of data acquisition and sample preparation.
Bioinformatics
These nodes include numerous tools for sequence alignment, blast search, and RNAi gene mapping, which can be linked to the output of other nodes.
Implementation
The architecture of HCDC-HITS was based mostly on Eclipse plug-in framework and Eclipse-KNIME 11 data workflow systems. HCDC-HITS is a functional node set, working together with the KNIME package. A plug-in for opening and processing proprietary HCS files (library, numeric results, and images) was developed within the KNIME environment. All of those open-source components (Eclipse environment, KNIME, R-Project, Weka, and ImageJ) were chosen for their platform independence, openness, simplicity, and portability. They are also the fastest pure Java image- and data-processing programs currently available. The programs have a built-in command recorder, editor, and Java compiler; therefore, they are easily extensible through custom plug-ins. The pipeline model of HCDC-HITS describes the exact behavior of the workflow when it is executed. The nodes of HCDC-HITS are designed on the following principles:
Resource type: The source of data can be data tables (library or image-processing results) or collection of high-level images familiar to the user or a single image. Software supports all image types, which are supported by ImageJ and ImageJ plug-ins.
Computation: Data flow pipelines dictate that each processor be executed as soon as its data inputs are available, and processors that have no data dependencies among each other can be executed concurrently. They are used for integrating data from different sources, data capture, preparation, and analysis pipelines and populating scientific models or data warehouses. Control flows directly dictate the flow of process execution, using loops, decision points, and so on.
Interactivity: Node execution could be wholly automatic or interactively steered by the user. Data flows are combined by simple drag-and-drop from a variety of processing units. Customized applications can be modeled through individual data subpipelines.
Adaptivity: The nodes and workflow design or instantiation can be dynamically adapted “in flight” by the user or by automatically reacting to changed environmental circumstances.
Modularity: Processing units and containers should not depend on each other to enable easy distribution of computation and allow for independent development of different image-processing algorithms.
Easy expandability: In HCDC-HITS, as in KNIME, it is easy to add new microscope, data analysis, image-processing software nodes, or views and distribute them through a simple plug-in mechanism without the need for complicated install/reinstall procedures. To achieve this, data processing consists of a pipeline of interconnected nodes that transport data.
Conclusion
In this article, we have presented HCDC-HITS, which is a visual programming language and package for HCS users. We have described its unique workflow-based framework, which has been aimed at speeding up, in a visual way, data management and data analysis tasks. The HCDC-HITS development environment allows the user to rapidly build data processes from existing components and services and monitor their execution in the form of visual programming. We have developed an integrated set of nodes for RNAi library handling, bioinformatics, microscopy image management, automatic layout of graphs, static-type checking, process compilation, execution profiling, analysis, and optimization. It features a powerful and intuitive user interface, enables easy integration of new modules or nodes, and allows for interactive exploration of analysis results or trained models. HCDC-HITS is an open-source project available at http://hcdc.ethz.ch. It is free to profit, nonprofit, and academic users.
Availability and Requirements
Project name: HCDC
Operating system: Platform independent
Programming language: Java
Other requirements: ImageJ library
Installation: http://hcdc.ethz.ch/index.php?option=com_content&view=article&id=1&Itemid=3
License: GNU General Public License, Version 3
Project Web page and download: http://hcdc.ethz.ch
Footnotes
Acknowledgements
We thank A. Vonderheit and M. Stebler for testing HCDC-HITS and for the development of many workflows.