
Abstract
15-Lipoxygenase-1 catalyzes the introduction of molecular oxygen into polyunsaturated fatty acids to form a lipid hydroperoxide. The authors have developed an assay for the detection of lipid hydroperoxides formed by human 15-lipoxygenase (15-LO) in enzyme or cellular assays using either a 96-well or a 384-well format. The assays described take advantage of the ability of lipid hydroperoxides to oxidize nonfluorescent diphenyl-1-pyrenylphosphine (DPPP) to a fluorescent phosphine oxide. Oxidation of DPPP yields a fluorescent compound, which is not sensitive to temperature and is stable for more than 2 h. The assay is sensitive toward inhibition and robust with a Z′ value of 0.79 and 0.4 in a 96- and 384-well format, respectively, and thus amenable for high-throughput screening. The utility of DPPP as a marker for 15-lipoxygenase activity was demonstrated with both enzyme- and cell-based assays for the identification of hits and to determine potency by IC50 determinations.
Keywords
Introduction
H
The enzyme 15-LO-1, or its rodent ortholog 12/15-LO, has been implicated in pro- as well as anti-inflammatory processes. 9,10 Ovalbumin-sensitized 12/15-LO null mice display reduced airway inflammation, reduced cytokine production, and less proliferation of airway epithelial cells after exposure of allergen. 11,12 There are also reports suggesting that 15-LO-1 is involved in pathological processes such as cytokine release from airway epithelial cells, 13 cardiac inflammation, 14 atherosclerosis, 15-17 Alzheimer’s disease, 18 Hodgkin’s lymphoma, 7 and latent insulin resistance and diabetic nephropathy. 19,20 Also, 15-LO-1 inhibitors have been shown to rescue neuronal cells from cell death induced by oxidative stress. 21 The importance of 15-LO-1 in several diseases has provoked numerous attempts to identify specific inhibitors, including screening of chemical compound libraries, virtual screening, and identification of natural products. 21-32 Especially interesting was the discovery of potent and cell-active 15-LO inhibitors that recently was described by Bristol-Myer-Squibb. 27
As part of our ongoing studies of the human 15-LO-1 enzyme, we have developed an enzyme- and a cell-based assay amenable for high-throughput screening (HTS). In the assay described herein, 15-LO-1 catalyzes the peroxidation of linoleic acid or arachidonic acid, resulting in the formation of a lipid hydroperoxide, which has the ability to oxidize the reagent diphenyl-1-pyrenylphosphine (DPPP;
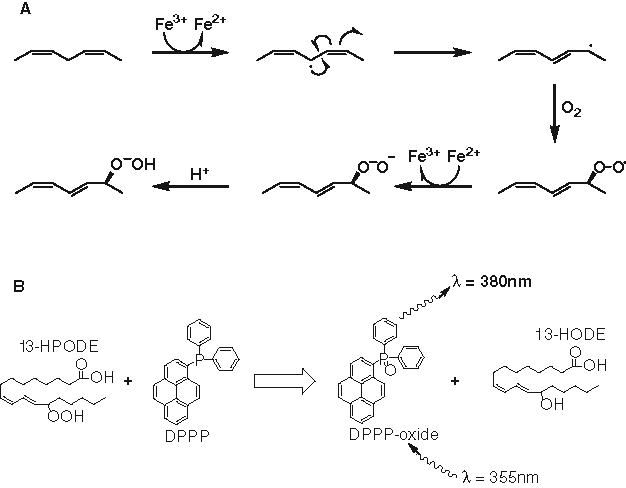
Oxidation processes. (
Materials and Methods
Materials
Arachidonic acid and linoleic acid were from NuCheck (Biomol GmbH, Hamburg, Germany) and diluted in ethanol to a final concentration of 100 mM and kept in glass vials at −20°C. High-performance liquid chromatography (HPLC) solvents were purchased from Rathburn Chemicals (Walkerburn, UK). 13(S)-Hydroperoxy-9(Z),13(E)-octadecadienoic acid (13-HODE), 13-HPODE, 15-HPETE, and 15-hydroxy-5(Z),8(Z), 11(Z),13(E)-eicosatetraenoic acid (15-HETE) were from NuCheck Prep Inc. (Elysian, MN, USA). DPPP was from Molecular Probes (Carlsbad, CA). Tissue culture medium, antibiotics, fetal calf serum, and Dulbecco’s phosphate-buffered saline (D-PBS) were from Gibco (Paisley, Scotland, UK). LOPAC (Library of Pharmaceutical Active Compounds) and all other chemicals were from Sigma-Aldrich (Stockholm, Sweden).
Expression and purification of human 15LO-1
Human 15LO-1 cDNA was amplified by PCR using the following primers that introduced cleavage sites for BglII and XhoI restriction enzymes at the 5′ and 3′ end, respectively: 15-LO-BglII 5′-ACC AGA TCT ATG GGT CTC TAC-3′ and 15-LO-XhoI 5′-TAT TCT CGA GTT AGA TGG CCA CAC TGT T-3′. Isolation of human eosinophils was performed as described in Feltenmark et al., 6 and total RNA was isolated from human eosinophils and used as a template after it was reverse transcribed. Subsequently, the coding sequence of 15LO-1, flanked by the restriction sites for BglII and XhoI, was subcloned into pCR4-TOPO, and thereafter the sequence was determined for both strands. The correct sequence encoding the full-length human 15LO-1 was cloned into pFastBac1 (Invitrogen, Carlsbad, CA) between the restriction sites BamHI and XhoI. Bacmid DNA was derived after transformation of DH10BAC bacteria with the pFastBac1 construct according to the manufacturer’s instruction. This bacmid DNA was used to transfect Sf9 insect cells using Cellfectin. Viral particles were collected 96 h posttransfection and amplified 3 times before a viral stock was obtained. The viruses were used to infect Sf9 cells cultured in Grace’s insect media supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin. Cells were harvested 48 or 72 h postinfection by centrifugation. Cell pellets were resuspended in homogenization buffer, 50 mM Tris-HCl (pH 7.5), 1 mM EDTA, and Complete Mini protease inhibitor cocktail and lysed by sonication. 15LO-1 was purified from the 20,000 g supernatant of homogenized Sf9 cells by metal affinity chromatography using a nickel column, followed by anion exchange chromatography using a RQ-6 column. Active fractions were pooled, desalted, and reinjected onto a RQ-1 column. Fractions with 15LO activity were pooled, aliquoted, and stored at −80°C until use. The 15LO-1 enzyme was approximately 90% to 95% pure as judged by Coomassie blue staining of sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and densitometry scanning of the gel.
15-LO-1 enzyme assay
15LO-1 activity was measured in black Optiplate-96 F NEW (#6005270, Packard BioScience, Meriden, CT), black Optiplate (Corning, Corning, NY), or black 384-plates from Corning (#3711) in triplicate or duplicate determinations as indicated. To each well in a 96-well plate, the following were added: 44 µL of 15-LO diluted in D-PBS at 3.18 µg/mL (2.8 µg/mL final concentration, corresponding to 0.14 µg of protein) or D-PBS alone and 1 µL of DMSO or compound dissolved in DMSO. This mixture was incubated for 5 min before the addition of 5 µL 2 mM linoleic acid. The plate was then incubated at room temperature for 10 min before the reaction was terminated with 2 volumes of methanol containing 0.125 mM DPPP. Fluorescence was measured after 30 min using either the Fusion™ Universal Microplate Analyzer from Packard BioScience (equipped with a quartz-halogen light source, blue PMT, excitation filter 330/30 nm, and emission filter 385/35 nm) or with a Spectramax Gemini fluorescence plate reader from Molecular Devices with an excitation wavelength of 363 nm and an emmission wavelength set to 380 nm. Detection was obtained from the top of the well for both instruments. Plates were processed by the use of a PlateTrak (PerkinElmer, Waltham, MA) or a Biomek FX (Beckman Coulter, Brea, CA).
Cell-based 15-LO-1 assay
The human Hodgkin lymphoma cell line L1236 was grown in a humidified atmosphere at 5% CO2 in RPMI1640 medium supplemented with 10% heat-inactivated fetal calf serum and L-glutamine. The cells were washed twice in D-PBS before they were aliquoted into wells. Test compound in DMSO or DMSO was added to each well, and the plate was incubated at room temperature for 10 min. Linoleic acid was added to yield a final concentration of 200 µM, and the plate was incubated for 30 min at room temperature. The reaction was terminated by the addition of 0.125 mM DPPP in methanol/DMSO (1:1 ratio), and the plates were incubated in darkness for at least 30 min before the fluorescence was measured as described above.
Analysis of lipoxygenase products by liquid chromatography
Plates were stored at −20°C for at least 60 min after the addition of methanol before centrifugation to remove precipitated proteins. Thereafter, monohydroxyacids were analyzed by reverse-phase HPLC (RP-HPLC) by injecting an aliquot of the supernatant onto a C18 NovaPak 3.9 × 150-mm column from Waters (Milford, MA) coupled to a Waters Alliance 2795 system or onto a Waters BEH C18 2.1 × 50-mm column coupled to an Aquity UPLC system. UV absorbance was measured at 235 nm. The retention time was compared with authentic 13-HODE standard, and occasionally, qualitative measurements were performed using a 2996 photodiode-array detector to verify the spectra.
Cytotoxicity assay
Membrane integrity was measured after incubation with or without test compound by determining the leakage of lactate dehydrogenase (LDH) using a commercially available kit (CytotoxOne, Promega, Madison, WI) according to the manufacturer’s instructions. Briefly, cell assays were performed as described above excluding the termination with methanol. Instead, plates were centrifuged, and the resulting supernatants were analyzed for LDH activity. Fluorescence was measured using the Gemini Spectramax instrument (Molecular Devices).
Determination of kinetic parameters and IC50 values
Kinetic parameters such as Km and Vmax as well as IC50 values were calculated by nonlinear regression using the GraphPad PRISM software (GraphPad, La Jolla, CA).
Results and Discussion
Cloning, expression, and purification of human 15-lipoxygenase-1
Human eosinophils isolated from whole blood were used to prepare RNA, which subsequently was reverse transcribed to cDNA. The coding sequence of 15-LO-1 was amplified by PCR as described. The 15-LO-1 cDNA was subcloned into pCR2.1-TOPO and sequenced to verify correct identity of the amplified fragment. The sequence obtained demonstrated 100% identity with the sequences deposited at the National Center for Biotechnology Information (NCBI). Interestingly, 1 clone retained 1 intron, leading to premature termination. Whether this is of any physiological significance remains to be determined. DNA from clones with correct sequences were cloned into either pFastBac or pFastBacHTb for expression of 15-LO-1 in Sf9 cells or in pET20b (for expression in Escherichia coli). 15-LO-1 protein expressed in Sf9 cells or bacteria was purified by ion metal exchange chromatography or ion exchange chromatography to approximately 95% homogeneity essentially as described previously.
33
A typical elution pattern from an ion exchange column is demonstrated in
Fluorescent detection of lipid hydroperoxides
Previous work has demonstrated that redox-sensitive dyes or fluorescent probes can be used to determine levels of lipid hydroperoxides.
29,34,35
Indeed, DPPP has proven to be a useful marker of lipid peroxidation and especially to monitor lipid hydroperoxides within cell membranes.
36,37
Whereas DPPP is essentially nonfluorescent, oxidation by a lipid hydroperoxide converts it to a phosphine oxide, which has fluorescence at 380 nm (
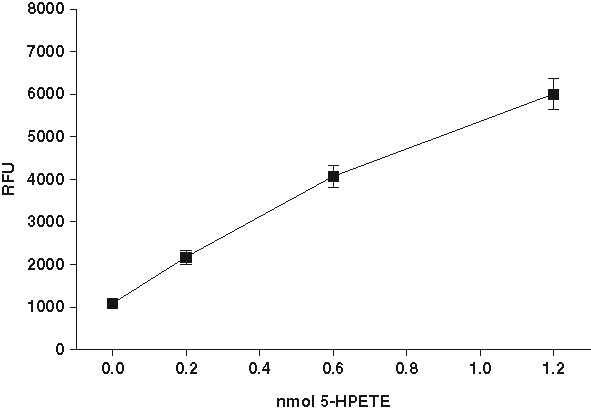
Fluorescence signal after oxidation of diphenyl-1-pyrenylphosphine (DPPP) by lipid hydroperoxide. (
Characterization of biochemical assay conditions and stability of fluorescent signal
Stability of purified human 15-LO-1 at −80°C was investigated by removing aliquots from the freezer at different time points. Enzyme activity was determined using the assay described in the Materials section, and the amount of 13-HPODE was determined by LC-UV detection. The results demonstrated that the enzyme was stable at −80°C for at least up to 90 days (Suppl. Fig. S2A). The stability of the purified enzyme at ambient temperature was investigated, and we observed a 30% reduction in enzyme activity after 2 h (Suppl. Fig. S2B). The enzyme kinetic parameters Km and Vmax were determined using oxidized DPPP as a marker of the amount of enzyme product (13-HPODE) or by direct quantification by RP-HPLC. The Km for the DPPP experiment is apparent (Km(app)) because we did not directly measure the amount of enzyme product. The Km(app) of linoleic acid was 30 µM for the fluorescent assay and 49 µM for the HPLC-based assay (Suppl. Fig. S3A-B), which is in line with previous reported values. However, several previous reports have used potassium linoleate as a substrate by diluting the linoleic acid in KOH in a 1:1 ratio. We determined the Km(app) of potassium linoleate, and it was 198 µM (Suppl. Fig. S3A). On the basis of these results, we draw the conclusion that the physical form of the fatty acid substrate (i.e., free fatty acid or an ionic form, such as potassium linoleate) can affect the Km(app) to a high degree. To ensure that we were screening at or above the Km(app), we chose 200 µM of linoleic acid as the substrate concentration for the screening assay. The formation of product was linear for up to 10 min when 0.3 µg of enzyme was used (Suppl. Fig S4A). The linearity of product formation versus the amount of enzyme was also investigated, and we observed a linear relationship for up to 0.35 µg of purified protein (Suppl. Fig. S4B). The DMSO tolerance was investigated using 0.14 mg of protein and an incubation time of 10 min. The results demonstrate that the inclusion of 1% DMSO (by volume) changed the enzyme activity by 10% in a significant manner (Suppl. Fig. S4C). Because of the rather limited effect on enzyme activity by 1% DMSO, we considered this reduction acceptable.
To determine the stability of the fluorescence of DPPP oxide, we incubated the DPPP-containing assay mixture for different time periods before measuring fluorescence. The results demonstrated that once the enzymatic reaction was stopped by the addition of methanol, both total (with enzyme) and basal (without enzyme) signal remained stable, indicating that DPPP oxide is stable for at least 120 min (Suppl. Fig. S5), meaning that several plates can be batch-processed and analyzed at a later stage and that accurate measurement of enzyme activity can be made anywhere from 15 to 120 min after addition of DPPP. The amounts of lipid hydroperoxide needed to oxidize were investigated by adding a fixed amount of lipid hydroperoxide to increasing amounts of DPPP in assay buffer containing 2 volumes of methanol to simulate assay conditions. It was evident that DPPP needed to be in excess to ensure that a majority of the lipid hydroperoxides were reduced to the corresponding mono-hydroxy acids (Suppl. Fig. S6).
Because the fluorescent signal for detection is based on a redox reaction and thus potentially sensitive to quenching by redox-active compounds, we investigated the effects of 2 known antioxidants on the enzyme activity by either fluorescent detection or HPLC detection of 13-HPODE. These compounds have previously been shown to possess an antioxidative effect,
38
and our aim was to determine if the oxidation of DPPP was affected by these compounds. As depicted in
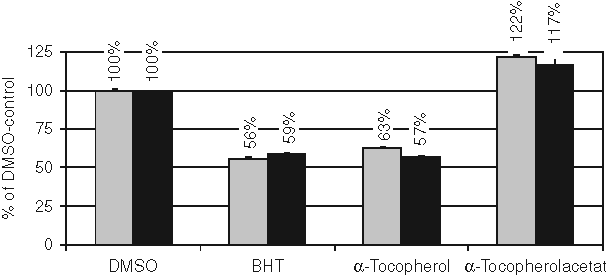
Effects of antioxidants on 15-LO-1 activity. The effects of antioxidants on 15-LO-1 activity were determined with both fluorescence and high-performance liquid chromatography (HPLC) analysis of the same sample. The activity was normalized to the DMSO vehicle control sample. Results are the mean ± SD (n = 3) from 1 representative experiment out of 2. BHT, butylated hydroxytoulene. Gray bars represent fluorescence detection, and black bars represent HPLC-based detection of 15-HPETE.
Determination of intraplate and interplate variability using the 96-well assay format
The results obtained so far (i.e., stability of DPPP oxide and correlation to HPLC values) indicate that the assay could be used for screening purposes to identify novel inhibitors. Thus, the assay was investigated further by studying the variability of the assay in 96-well plates. The intraplate and interplate variability was determined on 3 plates, using 32 total values (with 15-LO-1), 32 basal values (without 15-LO-1), and 32 inhibited values (with 15-LO and 2 µM of control inhibitor). Signal-to-background (S/B) values (mean of total signal/mean of basal [or inhibited] signal), percent coefficient of variation (%CV), and Z′ factors were calculated for each plate. The Z′ factors of the assay, representing statistical parameters for the quality of the assay (i.e., screening window coefficients), were calculated according to the formula of Zhang et al. 39 The S/B values for total (with 15-LO) over basal (without 15-LO) signal were acceptable for all 3 plates (between 6.9 and 8.4), and the %CV ranged from 5.5% to 10.1% for total signal. These data led to calculated Z′ values ranging between 0.61 and 0.77, indicating that the assay was robust enough for screening purposes.
Validation of the 96-well assay in HTS mode
The assay was validated in HTS mode by using 60 randomly selected 96-well plates (4800 compounds). Compounds were tested at 10 µM final concentration. Screen parameters (signal-to-noise ratio [SNR], %CV, and Z′ factors) were calculated for each plate. The following controls were included on each assay plate: (a) absence of 15-LO, (b) absence of inhibitor, and (c) presence of known inhibitor. The S/B values were calculated for total over basal signal. The data in
Summary of Assay Parameters
Results obtained from the 96-well assay in HTS mode and screening of a chemical compound library
Results from the validation demonstrated that HTS could be feasible using the DPPP method. Thus, a set of another 565 plates was screened using the same conditions as during the prescreen. The assay parameters for the HTS are presented in
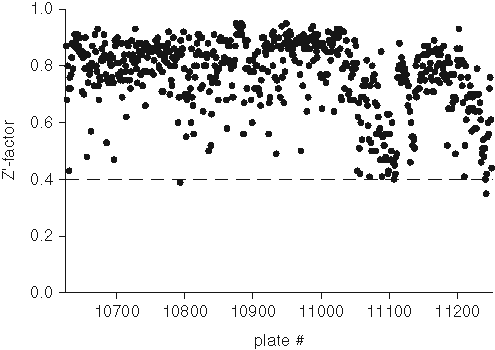
Z-factor plot from high-throughput screening (HTS) using the biochemical assay in 96-well plates. The Z′ factor from a total of six hundred twenty-five 96-well plates are represented by solid dots. The dashed line indicates the threshold of 0.4.
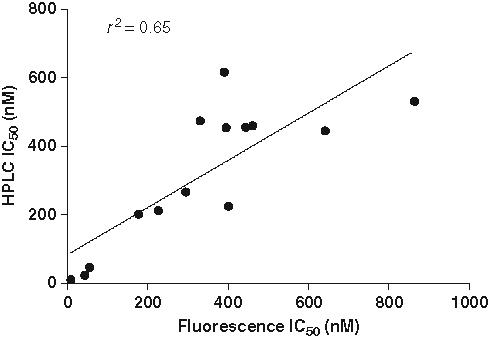
Correlation of IC50 values from diphenyl-1-pyrenylphosphine (DPPP)– and high-performance liquid chromatography (HPLC)–based methods. IC50 values for 14 compounds were determined using either the fluorescent method or the HPLC-based method. Assays were performed in a 96-well plate as described in Materials and Methods. The IC50 values are from a single experiment performed in triplicate.
Reformatting of the enzyme assay into a 384-well format and screening of a chemical compound library
To decrease the amount of reagents and enzyme, we reformatted the 96-well assay into a 384-well format with a final volume of 50 µL. However, the low-volume aspiration and dispensing of DPPP dissolved in methanol did not perform well with the 384-channel head. Therefore, DPPP was dissolved in a mixture of MeOH and DMSO at a 1:1 ratio. This new solvent for DPPP increased background fluorescence, and thus the 384-well format yielded an assay with Z′ values around 0.5. We investigated the usefulness of the 384-well format by screening the LOPAC library (1280 compounds) from Sigma-Aldrich. The sensitivity toward inhibitors and the robustness of the assay during this small screen were investigated. Furthermore, plates were analyzed with both fluorescent detection and with LC detection of lipid hydroperoxide.
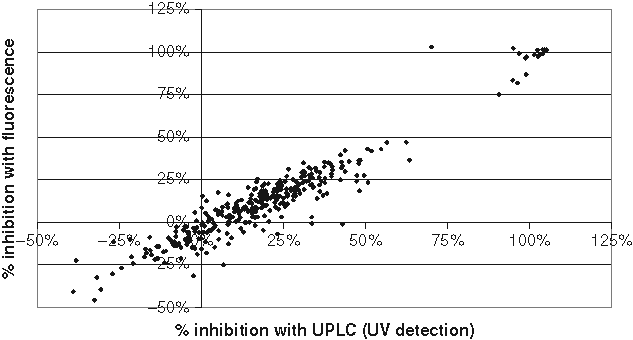
Correlation between diphenyl-1-pyrenylphosphine (DPPP)– and liquid chromatography (LC)–based screen of 15-LO inhibitors. Compounds from one 384-well plate were screened at 10 µM as described in Materials and Methods. The amount of 15-HPETE was quantified by either DPPP fluorescent detection or high-performance liquid chromatography (HPLC)–based detection. The results from both methods were plotted against each other, and the results are presented as percent inhibition. The results are from 1 representative plate out of 2, each screened in single points. Z′ values were 0.39 and 0.44 for the HPLC and DPPP assays, respectively.
To identify hits that acted as nonselective inhibitors, either as antioxidants or as iron chelators, we first determined the potency of the hits in the HPLC-based assay to avoid effects of the oxidation of DPPP (
Characterization of Hits
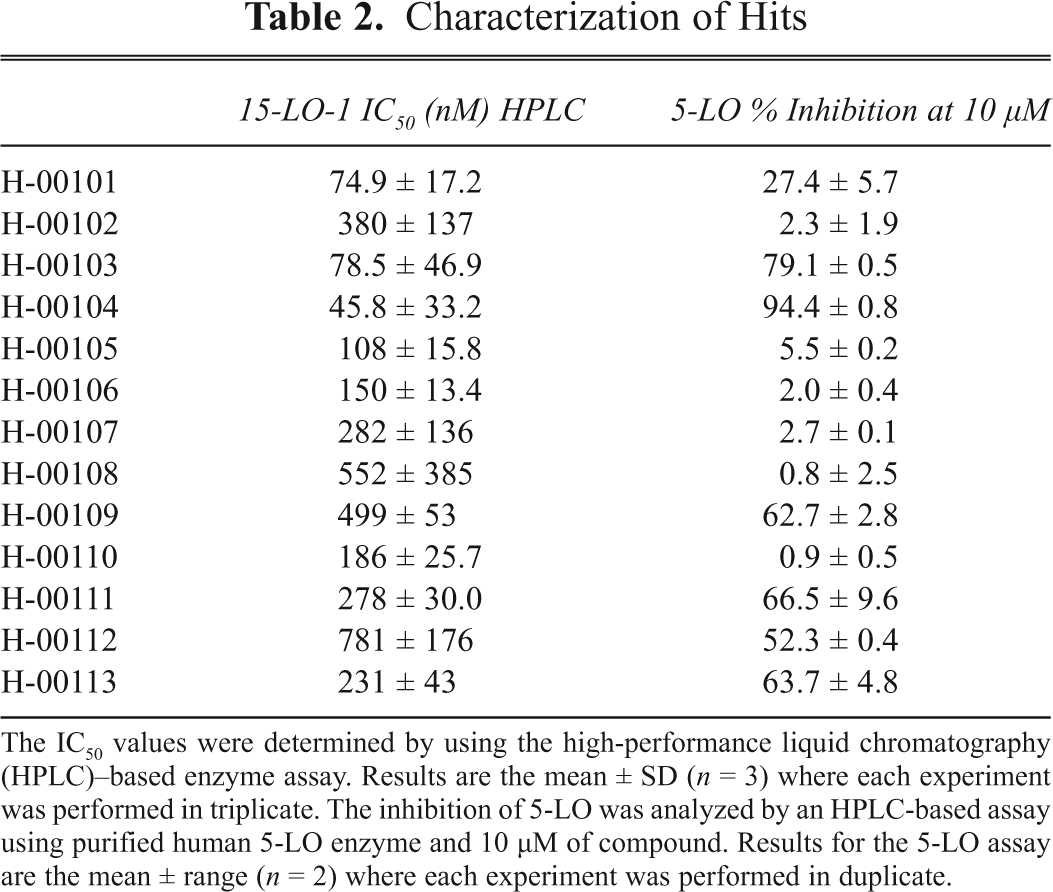
The IC50 values were determined by using the high-performance liquid chromatography (HPLC)–based enzyme assay. Results are the mean ± SD (n = 3) where each experiment was performed in triplicate. The inhibition of 5-LO was analyzed by an HPLC-based assay using purified human 5-LO enzyme and 10 µM of compound. Results for the 5-LO assay are the mean ± range (n = 2) where each experiment was performed in duplicate.
Overall, the biochemical HTS assay using DPPP as a marker of 15-LO-1 activity yielded 4.3% false positives (i.e., hits that could not be verified in the hit verification screen). The correlation between the percent inhibition of the confirmed hits in the primary screen and the IC50 values demonstrated a R 2 = 0.46. The major assay for filtering nonselective compounds was in this case the 5-LO enzyme-assay, which reduced the amount of hits by approximately 50%.
Development and validation of a cell-based assay in a 96- or 384-well format
The cell line L1236 was recently described to possess high endogenous expression of 15-LO-1, and the assay essentially has been described elsewhere.
7
We have used this cell assay to develop a microplate assay that is amenable for screening and for determination of inhibitory potency. To establish basic assay conditions, we titrated the amount of substrate and also investigated the linearity of product formation with respect to incubation time (Suppl. Figs. S7-S8). The final assay in a 96-well plate contained 50,000 cells per well and 12.5 µM of arachidonic acid. After incubating the cells with either DMSO or test compound, arachidonic acid was added to initiate 15-HPETE biosynthesis. The reaction was terminated after 10 min by the addition of 1 volume of methanol, and the amount of 15-HPETE was quantified by HPLC. The assay parameters, Z′ and %CV, are shown in
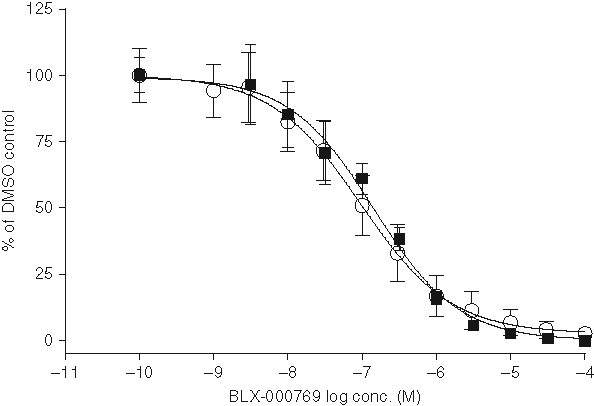
IC50 curve of BLX-000769 in the cell assay. IC50 curves of BLX-000769 were determined at 10 different occasions using the cell-based assay. All data points were normalized so that the DMSO control represents 100% activity. Results are the mean ± SE (n = 10 for high-performance liquid chromatography [HPLC] and n = 5 for diphenyl-1-pyrenylphosphine [DPPP]) where each experiment was performed in triplicate. Open circles demonstrate the IC50 curve obtained with HPLC detection. Solid squares represent the curve obtained with the DPPP assay. The IC50 values were determined by nonlinear regression to be 97.5 nM for the HPLC assay and 145 nM for the DPPP assay.
Conclusion
We have developed both enzyme- and cell-based assays amenable for the identification of novel 15-LO-1 inhibitors, based on a new application for DPPP. This fluorophore has been used to study lipid hydroperoxides in various samples, but it has not been used previously in screening applications. In this report, we demonstrate that oxidation of DPPP to a fluorescent molecule by lipid hydroperoxides yields a robust assay and accurately functions as a marker of lipoxygenase-derived lipid hydroperoxides. Furthermore, the DPPP-based detection of lipid hydroperoxides shows a very good correlation to the amount quantified by HPLC. The DPPP assay could be scaled down successfully from a 96-well format to a 384-well format, thus making the assay more cost-effective. Both the 96- and the 384-well formats were used to identify inhibitors as well as for determining potency by analyzing IC50 values. The major advantage of DPPP, as compared with colorimetric redox-sensitive dyes, 27 is that DPPP can be used directly in cell-based assays, both for screening purposes as well as for lead optimization. The DPPP-based detection used herein is in contrast to a previously described chemiluminescent method for detecting lipid peroxides, 40 not dependent on a secondary enzymatic step involving peroxidases. Thus, the assay described herein is probably more suitable for studying inhibitors.
The discovery of potent and cell-active 15-LO-1 inhibitors as described herein and in Weinstein et al. 27 might lead to better pharmacological tools to study the physiological and pathological role of 15-LO-1. Further studies using 15-LO-1 inhibitors might reveal novel functions for this enzyme in a similar manner as it was described to play a role in cell death in both neurons and oligodendrocytes. 21 In summary, we have used enzyme and cell-based 15-LO-1 assays to successfully identify new potent 15-LO-1 inhibitors for the further development into unique lead series.
Footnotes
Acknowledgements
Prof. Hans-Erik Claesson is gratefully acknowledged for carefully reviewing the manuscript.
This work was financially supported by Orexo AB and by the European Commission FP6 Grant LSHM-CT-2004-005033. The report reflects only the authors’ views, and the European Commission is not liable for any use that may be made of the information herein.