
Abstract
Current methods for high-throughput screening (HTS) use a serial process to evaluate compounds as inhibitors toward a single therapeutic target, but as the demand to reduce screening time and cost continues to grow, one solution is the development of multiplex technology. In this communication, the multiplex assay capability of a mass spectrometry (MS)–based readout system is verified using a kinase and esterase reaction simultaneously. Furthermore, the MS-based readout is shown to be compatible with a typical HTS workflow by identifying and validating several new inhibitors for each enzyme from a small library of compounds. These data confirm that it is possible to monitor inhibition of multiple therapeutic targets with one pass through the compound repository, thus demonstrating the potential for MS-based methods to become a method of choice for HTS of isolated enzymes.
Keywords
Introduction
O
The idea of using mass spectrometry for multiplex enzyme assay screening is not new; in fact, Hsieh et al. 9 showed the potential for a MALDI-MS-based readout for evaluating inhibitors to several enzymes simultaneously. In this proof-of-concept study, they clearly showed that up to 3 enzyme reactions could be monitored simultaneously with a simple MALDI-TOF instrument; however, screening quality issues, including reproducibility, precision, false hits, and validation, were not addressed. Similarly, Liesener et al. 10 demonstrated the multiplex potential by using ESI-MS to screen for multiple proteolytic activities. In this study, the investigators used 5 specific peptide substrate/product pairs to evaluate the types of protease activities present in fractionated snake venom. Furthermore, mass spectrometry has been used for many years in clinical settings to simultaneously evaluate multiple enzymes in cases such as enzyme deficiency syndromes from dried blood drops from newborns. 11 Multiplex assays to evaluate cytochrome P450 inhibition in liver microsomes have been also reported using an liquid chromatography/tandem mass spectrometry (LC/MS/MS)–based assay. 12 However, in each of these latter cases, the measurements were targeted only at determining if the enzyme activity was present or if a specific drug compound inhibited one of the targeted enzymes, and they were not involved with evaluating a compound repository for specific inhibitors of the enzyme activities. As a result, the concept of using an MS-based readout for multiple enzymes was demonstrated but not expanded into the context of practical utility for a high-throughput inhibitor screening platform. Thus, the potential for multiplex assays using an MS-based readout is apparent, but additional integration and implementation of these methods are needed to validate the methodology and include it in an HTS workflow.
Recently, we have demonstrated the utility of a MALDI triple-quadrupole (QqQ)–MS readout for HTS applications with speeds reaching 1 sample per second, a robust readout with Z′ values >0.5, and a hit confirmation rate at 100%. 8 In this application note, the high-throughput MALDI-QqQ-MS-based workflow is evaluated for its multiplex capabilities using 2 unrelated enzymes, acetylcholinesterase (AChE) and cyclic AMP-dependant protein kinase A (PKA). The data presented demonstrate not only the feasibility of the multiplex reaction but also how a MALDI-QqQ-MS readout is well suited for integration into all aspects of an HTS workflow from the primary screen against multiple enzymes without interference, to hit validation and into lead optimization. The net result is a readout system that produces better quality data in a multiplex format than is currently available using existing HTS readout methods.
Materials and Methods
Materials
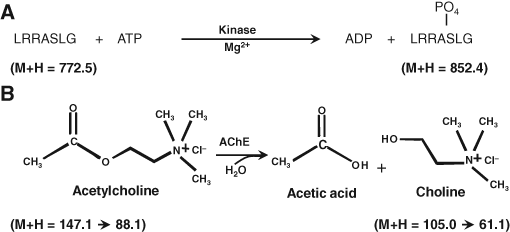
Acetylcholinesterase from Electrophorus electricus, acetylcholine, choline, α-Cyano-4-hydroxycinnamic acid (4-HCCA), PKA inhibitors (staurosporine, Ro-31 8220, and GF109203X), and AChE inhibitors (BW284c51, tacrine hydrochloride, (±) Huperzine A) were purchased from Sigma-Aldrich (St. Louis, MO). The catalytic subunit of cAMP-dependent protein kinase (PKA, V5161) and Kemptide (V5601) were purchased from Promega (Madison, WI). Ammonium monobasic phosphate and formic acid were from EMD Biosciences (San Diego, CA) and J. T. Baker (Phillipsburg, NJ), respectively.
Equipment
A Beckman Coulter Multimek NX (Fullerton, CA) liquid handling robotics system equipped with a 384-well low-volume transfer head was used for preparing enzymes, compound dilutions, substrates/adenosine triphosphate (ATP), and matrix solution plates and assay reactions.
Multiplex (PKA and AChE) assay
Individual and multiplex enzyme assays were prepared using PKA with Kemptide substrate and AChE with acetylcholine substrate. The reactions were initiated by transferring and mixing 5 µL of inhibitor compound from 384-well plates (prepared in the UC Drug Discovery Center, Cincinnati, OH) to an assay plate containing 20 µL of enzyme immediately followed by the transfer of 25 µL of the substrate to the assay plate to start the reaction. All assays were conducted in 40 mM Tris:HCl, pH 7.5, containing 20 mM MgCl2, 0.1 mg/mL bovine serum albumin (BSA). For reactions containing PKA (either individually or in combination), 0.6 units/well of PKA, 5 µM ATP, and 0.5 µM Kemptide were also included into the reaction mixture. For reactions containing AChE (either individually or in combination), 0.003 units/well of AChE enzyme and 25 µM acetylcholine were also included into the reaction mixture. After 15 to 30 min, the reactions were quenched by diluting 5 µL of the reaction mixture into 45 µL of matrix solution (60% acetonitrile containing 0.1% formic acid, 5 mg/mL 4-HCCA matrix, and 10 mM ammonium phosphate monobasic), and then a 1- to 2-µL aliquot from the quenched reaction plate was deposited onto target plates with no intervening sample cleanup step. After the samples were allowed to dry, the target plates were transferred manually to the MALDI-QqQ-MS system for analysis.
MALDI-QqQ-MS Readout
The MALDI-QqQ-MS data were collected on an Applied Biosystems/MDS Analytical Technologies FlashQuant™ system consisting of a FlashLaser™ source coupled with an API 4000 QTRAP® mass spectrometer system. The system was operated in positive ion mode at unit resolution with multiple-reaction monitoring (MRM) as the readout. For the AChE reaction, the MRM transitions were optimized for 147.1/88.1 for acetylcholine and 105.0/61.1 for choline with dwell times of 5 ms each and a total duty cycle of 20 ms. Collision energies were 11 eV and 26 eV for acetylcholine and choline, respectively. For the PKA readout, limited interference from the matrix background at this mass range produced an optimal signal to background using a precursor-to-precursor transition for the Kemptide substrate at m/z 772.5 and the p-Kemptide product at m/z 852.4, with dwell times of 5 ms each and a total duty cycle of 20 ms, and thus a full MRM transition was not necessary for this substrate/product pair. The collision energies for these precursor-to-precursor transitions were optimized at 6 eV. The MALDI-MS detection levels for the substrates and products for these reactions were optimized to 25 nM for the PKA reaction and 1.25 µM for AChE reactions. Although lower limits of quantitation (LLOQ) were achieved by MALDI-MS (LLOQ for choline at 20 nM), the use of inexpensive, label-free reagents allows for the enzyme and substrate concentrations to be adjusted to any desired level to ensure sufficient signals and a robust readout without needing maximum sensitivity as discussed previously. 4 For all readouts, the laser was fixed at 1000 Hz, and the raster speed across the MALDI sample plate was set to 4.5 mm/s (or 1 sample per second across each row of a 384-well plate). However, due to the plate movement time from row to row and the time to load each new plate into the system, the total time to load and scan a 384-well plate of samples in full MRM mode in the multiplex format is less than 1.5 s/sample, which is under 10 min per plate. More details regarding the tuning, effect of scan speed, and the optimization of parameters for PKA and AChE readout reactions are available in previous reports. 6,8
Compound library screening
A small library of 1008 compounds (3 × 384-well plates) diluted for a final concentration of 5 µM in each reaction well was used to screen for inhibitor activity, in triplicate, as the primary screen. Primary screening results are represented as percent maximal enzyme activity as described previously. 8 The most potent compounds (showing more than 40% inhibition) from the PKA and AChE primary screen were selected for secondary screening. IC50 curves from 10-point concentration-dependent inhibition assays were generated and plotted to establish the rank order of inhibitory potency.
Data analysis
The MRM peak areas for the substrates and products were used to calculate percent conversion of substrate to product (% C) and the degree of inhibition as percent maximal enzyme activity (% MA), as previously reported. 6,8 Percent MA was used to plot and calculate IC50 values from 10-point concentration-dependent inhibition curves using the GraphPad Prism 5.0 program (GraphPad Software, Inc., San Diego, CA), based on a sigmoidal dose response. Data are presented as the mean ± SD from a minimum of 3 independent experiments. Z′ values were also calculated as previously described. 13
Results and Discussion
To test whether inhibitors for 2 enzyme reactions could be monitored simultaneously without interference, we prepared reactions using PKA and AChE either individually or in combination (
Enzyme reactions for the multiplex assay evaluation. (
To further validate that the combination assays would not interfere with inhibitor screening for either of the target enzymes, we generated 10-point dose-dependent inhibition curves for each of the enzymes individually and in the multiplex format. For these studies, commercially available inhibitors for PKA (staurosporine, Ro-31-8220, and GF109203X) and AChE (BW284c51, tacrine hydrochloride, and (±) Huperzine A) were used and IC50 values calculated. As demonstrated in
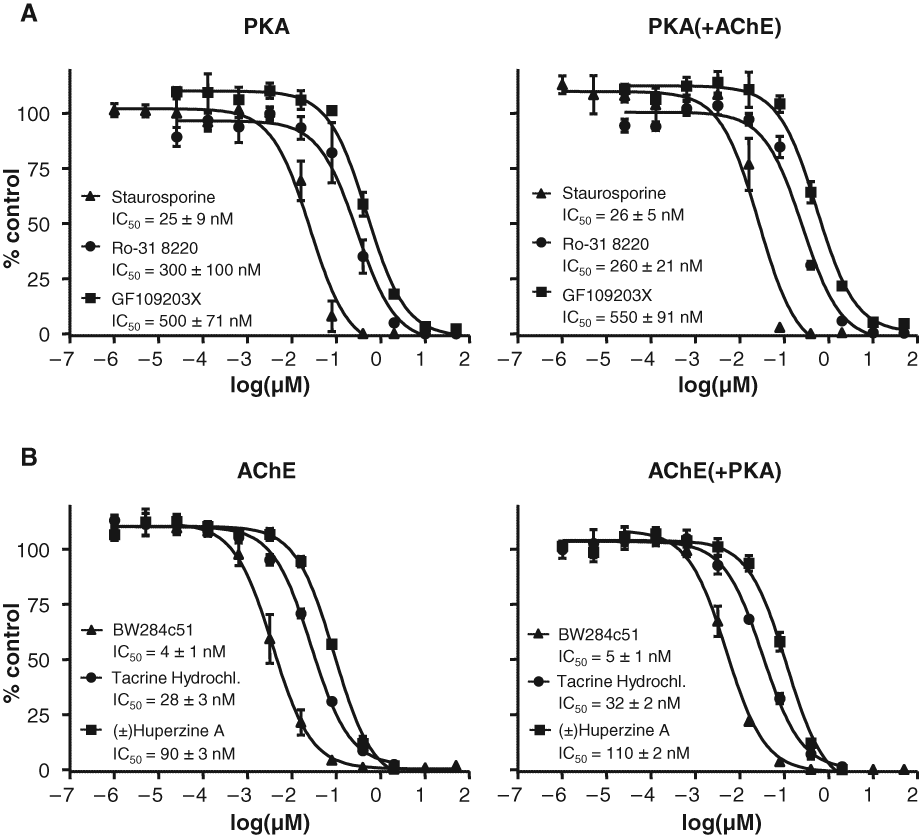
Effect of the multiplex enzyme reactions on known inhibitors of protein kinase A (PKA) and acetylcholinesterase (AChE). Ten-point dose-dependent inhibition assays were set up to evaluate whether the combination of enzymes would change the IC50 values for commercially available PKA and AChE inhibitors. The inhibitor assays were done in triplicate with the results plotted as the rank order of inhibitor potency for a series on known inhibitors, as indicated. The IC50 values are listed as the mean ± SD for each inhibitor compound. (
To validate the multiplex MALDI-QqQ-MS readout in a high-throughput work flow, we evaluated a small library of 1008 structurally diverse compounds across three 384-well microplates in triplicate at a single dose (5 µM) in a combination reaction containing both PKA and AChE. The reaction plate also included a series of positive and negative control reactions. As expected, all of the known inhibitors of PKA and AChE resulted in complete inhibition of the respective enzyme activity (data not shown). For the library of compounds, data were plotted as percent maximal enzyme activity (% MA) from the average of triplicate wells (
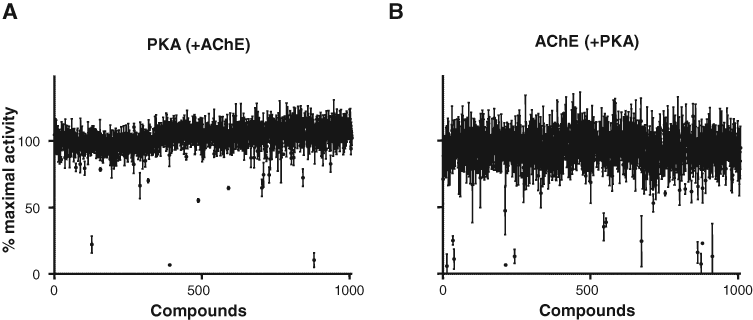
Primary screening results from a multiplex reaction. A small library of 1008 structurally diverse compounds was assayed in triplicate for inhibitory activity at a single dose of 5 µM each in a multiplex format containing protein kinase A (PKA) and acetylcholinesterase (AChE). The mean and standard deviation of the percent maximal activity are plotted for each compound. (
To confirm the hits detected from this small primary screen, we selected compounds producing less than 60% enzyme activity (more than 40% inhibition) for further validation in dose-dependent inhibitions assays.
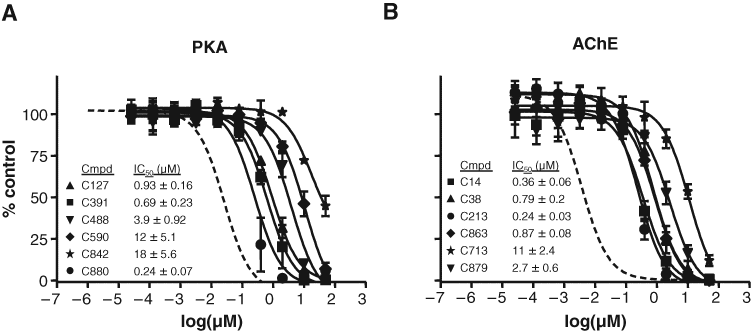
Secondary screening for inhibitor validation. Each compound from the primary screen that produced greater than 40% inhibition at the single concentration of 5 µM was subjected to validation in triplicate 10-point inhibition assays against protein kinase A (PKA) or acetylcholinesterase (AChE). IC50 curves for each of the compounds as the mean ± SD. (
Collectively, these data support the conclusion that a multiplex approach to inhibitor screening is indeed feasible in an HTS workflow from primary screening through hit validations and rank order of inhibitory potency. These multiplex capabilities coupled with the other advantages of MS-based screening applications, which include label-free (and thus inexpensive) reagents, minimal or no false-positive readouts, and robust analytical precision, all suggest that MS-based screening readout may have a bright future if it is successfully adopted and integrated by the screening community.
There are, however, some considerations that must be taken into account during the assay development phase for the successful implementation of multiplex readout by MS. First, one obvious observation is that the enzymes to be evaluated in a multiplex format must be compatible under that same reaction conditions. This would include all of the typical enzyme parameters (e.g., km of substrates and cofactors, pH, linear reaction phases) that are typically necessary for the individual reactions. Furthermore, concerns about cross-reactivity of enzymes with multiple substrates have to be considered. For example, a multiplex assay to evaluate multiple kinases could be problematic without very specific substrates for each. Similarly, one has to be careful not to combine a protease with another enzyme with a peptide substrate that is also hydrolyzed by the protease. Second, any MS-based readout assay requires that the analytes to be measured can be ionized and detected in the mass spectrometer. Although it is expected that there will be some substrates and products that will not ionize sufficiently to effectively use an MS-based readout, to date we have not encountered specific enzyme classes that have caused an issue, 5 although a broader scope of enzyme classes still needs to be evaluated. Although each of these issues can be readily addressed during the assay development phase by including the appropriate control reactions, they are worth noting so that they can be avoided from the outset.
To summarize, data presented here indicate that the MALDI-QqQ-MS readout has the selectivity to accurately measure multiple enzyme reactions in a single mixture in a typical HTS workflow—from primary screen through hit validation and rank order of inhibitory potency. With the clear advantages of label-free readout, excellent analytical precision, reproducibility, and the ability to screen for inhibitors of multiple therapeutic targets simultaneously, MS-based readout has the potential to transform how we evaluate isolated enzymes in the future. In this context, the ability to measure multiple substrates and products simultaneously by MALDI-QqQ-MS also opens the possibility not only to conduct 2 assays together in a single well but also to evaluate 3 or more targets together, thus further reducing the screening time and consumables.
Conceptually, the speed and cost advantages of using a multiplex approach for screening several different therapeutic targets simultaneous are obvious, but other enzyme combinations could also be evaluated as a means of refining compound classes even at the primary screening level. For example, it may be possible to screen a therapeutic target concurrently with related enzymes for which inhibition needs to be avoided. Similarly, one could concurrently include a control enzyme screening reaction to select against non-mechanistic-based or the so-called promiscuous inhibitors 15 that disrupt a variety of enzyme activities. In either case, the hits from the primary screen could be selected based on the inhibition against the therapeutic target and the lack of inhibition toward the other control enzymes activities included in the multiplex reaction.
In conclusion, the goal of this report was to demonstrate that multiplex enzyme assays were feasible using the MALDI-MS-based readout and to provide data indicating that the approach was amendable to a typical HTS workflow from primary screening through hit validation and rank order of inhibitor potency. The data presented clearly show that multiplex assays can be performed without interference, that new inhibitors for each enzyme can be identified, and that the process is amenable to a typical HTS workflow, albeit with a very small test library of compounds. The next logical question is how this process might scale to a much larger library to compete with current HTS assays. From a throughput standpoint, each plate can be run in under 10 min by MALDI-MS readout, from loading of one plate to loading of the next, thus corresponding to a practical rate of about 1.5 s/sample on a 384-well plate. Given that the enzyme assays take about 15 to 30 min to run, the rate-limiting step in the process (like most other HTS readout assays) is the time it takes for the enzyme reaction to complete rather than the assay readout time. Nonetheless, additional improvements in the readout speed would have a significant benefit to the overall process. To that end, we have already reported assay speeds of less than 2 min per plate on a 384-well platform using the MALDI-MS readout for single enzyme assays 6 ; however, additional validation is needed to apply these same readout speeds to multiplex assays. Further considerations that still need to be addressed to move the process into a full HTS platform include automation and integration of the enzyme assay step with the mass spectrometry readout, as well as automated capture and processing of the output data. In the current state, the MALDI-MS-based readout system is run in a stand-alone format with plates generated by liquid-handling robotics that are manually transferred to the instrument. Current development is targeted at integrating a plate-loading system to stack and run the target plates continuously on the MALDI-MS in an unattended manner. Furthermore, to improve the automation and begin to integrate the enzyme assay step with the mass spectrometry readout, we have also begun to adapt our enzyme assay reactions from the Beckman-Coulter NX platform to a fully automated Evotec (PerkinElmer, Waltham, MA) HTS system, thus moving toward a true high-throughput workflow. Similarly, additional integration and automation of data analysis needs to be implemented. To date, we have demonstrated that the substrate/product output data from the mass spectrometer can be directly imported into the Genedata software used by the HTS group within our Drug Discovery Center. Once the data are imported into this system, we have shown that all the typical visualization, statistics, and data management tools available in that system can be used to evaluate and present the MALDI-MS data. Further integration and implementation of this process will also be necessary to develop fully automated HTS workflow. Nonetheless, without demonstrating that the MALDI-MS readout assay has considerable advantages over existing assay formats, developing the fully automated and integrated system would not be practical. In this report, we present the advantages of MALDI-MS-based screening with a robust multiplex readout and no false-positive hits as sufficient evidence to push forward into full HTS integration as described above. In the end, as pharmaceutical companies and contract research organizations continue to look toward technologies to reduce cost while maintaining the quantity and speed of the current drug discovery paradigm, this multiplex approach has the potential of transforming the way the HTS community performs isolated enzyme inhibitor screening in the future.
Footnotes
Acknowledgements
The authors thank Mike Wyder and Wendy Haffey for technical assistance and Sandra Nelson for her thoughtful discussions and screening advice.
This work is supported by a postdoctoral fellowship from MDS Analytical Technologies and the University of Cincinnati Millennium Scholars Fund.