
Abstract
Studies of the phosphodiesterase PDE7 family are impeded by there being only one commercially available PDE7 inhibitor, BRL50481. The authors have employed a high-throughput screen of commercial chemical libraries, using a fission yeast-based assay, to identify PDE7 inhibitors that include steroids, podocarpanes, and an unusual heterocyclic compound, BC30. In vitro enzyme assays measuring the potency of BC30 and 2 podocarpanes, in comparison with BRL50481, produce data consistent with those from yeast-based assays. In other enzyme assays, BC30 stimulates the PDE4D catalytic domain but not full-length PDE4D2, suggesting an allosteric site of action. BC30 significantly enhances the anti-inflammatory effect of the PDE4 inhibitor rolipram as measured by release of tumor necrosis factor α from activated monocytes. These studies introduce several new PDE7 inhibitors that may be excellent candidates for medicinal chemistry because of the requirements for drug-like characteristics placed on them by the nature of the yeast-based screen.
Introduction
C
Enzymes of the PDE4, PDE7, and PDE8 families are cAMP specific and are expressed by 4, 2, and 2 genes respectively. Rolipram, the first PDE4-specific inhibitor, has been used to demonstrate that inhibition of PDE4 can reduce inflammation, enhance cognition, inhibit HIV infection and replication, and induce apoptosis in chronic lymphocytic leukemia (CLL) cells, thus demonstrating a wide range of biological roles for this enzyme family. 1-3,5 Much less is known about the more recently discovered PDE7 and PDE8 enzymes, in part because of the limited availability of family-specific inhibitors. Although PDE7 is a potential target for treatment of inflammation, 6 only 1 PDE7 inhibitor BRL50481, 7 is commercially available, and this compound is significantly less active against PDE7B than it is against PDE7A (see Results). Because most PDE inhibitor discovery is performed through the structural characterization of the active site, combined with the novel synthesis and testing of compounds from a particular structural class, the compounds identified in these studies are generally not readily available to the research community. 8,9 Even fewer tools are available to study the postgenomically identified PDE8A and PDE8B enzymes, with the broad-spectrum PDE inhibitor dipyridamole being used for PDE8 inhibition. 1
The fission yeast Schizosaccharomyces pombe monitors extracellular glucose by a cAMP signaling pathway. 10 High glucose levels detected by a putative G-protein-coupled receptor, Git3, lead to adenylate cyclase activation via the Gpa2-Git5-Git11 heterotrimeric G-protein. Adenylate cyclase produces a cAMP signal that activates protein kinase A (PKA), which negatively regulates transcription of genes involved in gluconeogenesis and sexual development. Most of the genes of the PKA pathway have been identified in genetic screens that use a fusion of the PKA-repressed fbp1 + gene to the ura4 + gene of the uracil biosynthetic pathway. Defects in cAMP signaling lead to constitutive fbp1-ura4 expression, allowing growth on glucose-rich medium lacking uracil while conferring sensitivity to the pyrimidine analog 5-fluoro orotic acid (5FOA). 11 Screens for suppressors of this low PKA phenotype, which restore 5FOA-resistant growth, identified mutations in the cgs1 + and cgs2 + genes that encode the regulatory subunit of PKA and the cAMP PDE, respectively. 12,13
The growth phenotypes conferred by the fbp1-ura4 fusion were further exploited to develop a high-throughput screening (HTS) platform for PDE inhibitors by replacing the cgs2 + PDE gene with mammalian PDE genes and screening for compounds that confer 5FOA-resistant growth to strains that possess defects in glucose signaling. 14 Our initial screens, which focused on the PDE4 family, showed that the furanocoumarin imperatorin is a PDE4B inhibitor, thus explaining a prior study that demonstrated its ability to inhibit HIV replication, 15 an outcome also observed with PDE4 inhibition by rolipram. 5 Additional PDE4 inhibitors will be described elsewhere.
This current study describes the development and deployment of a PDE7A inhibitor screen and the identification of compounds that inhibit PDE7A and PDE7B activity. Foremost among these is BC30, which displays potent synergy with rolipram in inhibiting tumor necrosis factor α (TNFα) release by lipopolysaccharide (LPS)-treated U937 cells. Although the mechanism of PDE7 inhibition by BC30 remains to be determined, it appears to act at a site other than the cAMP binding site, as suggested by its unusual ability to stimulate cAMP hydrolysis in an in vitro reaction that contains the catalytic domain of the PDE4D enzyme, which displays ~60% similarity to PDE7A and PDE7B catalytic domains. These studies demonstrate the utility of our screening platform for the discovery of novel PDE inhibitors.
Materials and Methods
Yeast strains, media, and growth conditions
Strains CHP1189 (h+ fbp1-ura4+ ura4::fbp1-lacZ leu1-32 gpa2Δ pap1Δ cgs2::PDE7A1), CHP1209 (h− fbp1-ura4+ ura4::fbp1-lacZ leu1-32 git3Δ pap1Δ cgs2::PDE7B1), CHP1262 (h− fbp1-ura4+ ura4::fbp1-lacZ leu1-32 gpa2Δ pap1Δ cgs2::PDE4A1), CHP1268 (h− fbp1-ura4+ ura4::fbp1-lacZ leu1-32 git3Δ pap1Δ cgs2::PDE4B2), and CHP1186 (h+ fbp1-ura4+ ura4::fbp1-lacZ leu1-32 git11Δ pap1Δ cgs2::PDE4D3) were grown in YEA-rich or EMM-defined media as previously described. 13
Construction of S. pombe strains that express human PDE4 and PDE7 enzymes
The human PDE4A1 (Genbank accession number U68532), PDE4B2 (Genbank accession number L20971), PDE4D3 (Genbank accession number U50159.1), PDE7A1 (Genbank accession number NM_002603), and PDE7B1 (Genbank accession number NM_018945) open reading frames were PCR amplified using oligonucleotides that provide targeting sequences to the S. pombe cgs2 PDE gene locus. As previously described, 14 the PCR products were integrated by homologous recombination into the cgs2/pde1 locus, which had been disrupted with ura4+ to allow detection by 5FOA counterselection. 16 Subsequent crosses were carried out to introduce fbp1-lacZ and fbp1-ura4 reporters, along with mutations in the glucose/cAMP pathway and a deletion of the pap1+ transcriptional activator gene to enhance compound sensitivity. 17
HTS and 5FOA growth assays
High-throughput screens were performed at the Broad Institute’s Chemical Biology Program screening facility. Prior to screening, each HTS was optimized as follows. First, various mutations with respect to cAMP production were introduced into the strain background to determine whether they confer significant 5FOA sensitivity, a reflection of low basal cAMP levels.
10
Second, we determined the optimal concentration of cAMP that must be present in the culture prior to the HTS to repress the fbp1-ura4+ reporter that controls the 5FOA growth phenotype. If the reporter is not repressed prior to the screen, cells will be 5FOA sensitive because of the preexisting Ura4 enzyme. Finally, we determined the optimal concentration of cells to inoculate into the microtiter dishes to produce an HTS with a high Z factor (see below). For the PDE7A HTS, strain CHP1189 (gpa2Δ PDE7A) was cultured to exponential phase in EMM complete medium containing 2.5 mM cAMP to repress fbp1-ura4 transcription. Cells were collected by centrifugation and resuspended in 5FOA medium,
11
and 25 µL was transferred to 384-well microtiter dishes (untreated, with flat clear bottoms) that contained 25 µL 5FOA medium plus 100 nL of compounds (stock solutions were generally 10 mM). The starting cell concentration was 1 × 105 cells/mL. Positive control wells contained 5 mM cAMP in the 5FOA medium. Cultures were grown for 48 h at 30°C while sealed in an airtight container with moist paper towels to prevent evaporation. Optical densities (OD600) of the cultures were determined using a microplate reader to measure growth. Bioinformatic analysis of the results to determine composite Z scores was performed as previously described.
18
The Z factor of an assay is determined by multiplying the sum of the standard deviations of the positive and negative controls by 3, dividing by the absolute difference in the means of the positive and negative controls, and subtracting from the number 1. An assay with a Z factor of greater than 0.5 is considered sufficiently robust for HTS. Within a screen, individual wells are assigned a Z score, which represents the number of standard deviations above or below the mean of the negative control wells in that same assay plate. Duplicate Z scores for each compound are plotted onto a grid (
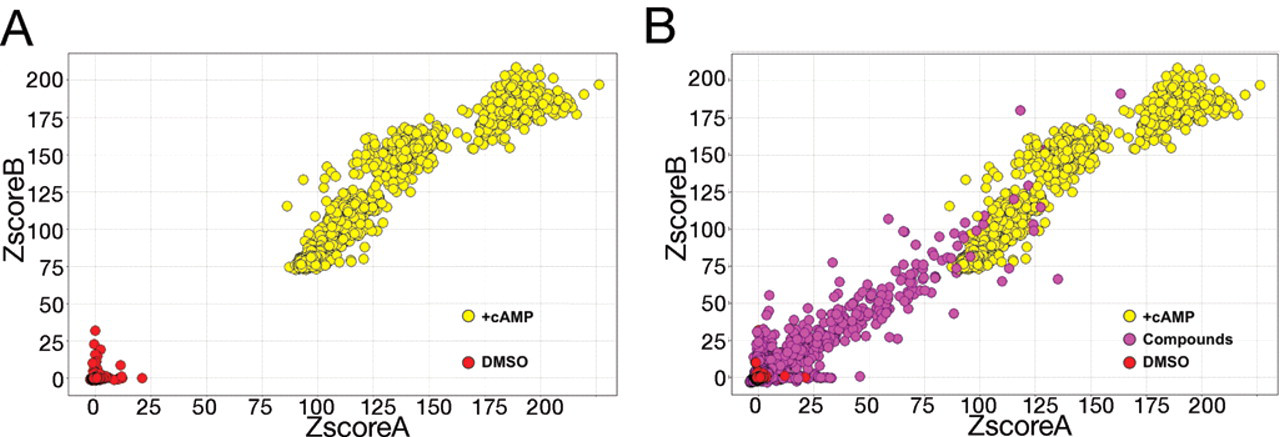
High-throughput screening data summary. (
In vitro enzyme assays
In vitro PDE assays were carried out as described by Wang et al., 19 in which the amount of cAMP remaining in a reaction is measured by scintillation counting after differential Ba(OH)2 precipitation of the 3H-AMP produced by hydrolysis from the unreacted 3H-cAMP. PDE4D (both catalytic domain and full-length PDE4D2) and PDE7B enzymes were generously provided by Drs. H. Wang and H. Ke; PDE7A enzyme was purchased from BIOMOL International (Plymouth Meeting, PA). Assays were conducted under conditions that resulted in less than 25% hydrolysis of cAMP in the absence of inhibitor. The rate of hydrolysis, which was constant over the 15-min duration of the reaction, was proportional to enzyme concentration. The initial cAMP concentration, 25 nM, was slightly below the published Kms for cAMP of 100 to 200 nM for PDE7A and 30 to 70 nM for PDE7B, 1 as well as our observed Km for PDE7B of ~50 nM, which was obtained using Michaelis-Menten kinetics analyzed by Lineweaver-Burke and Eadie-Hofstee plots (not shown). The IC50 is the concentration of inhibitor that reduces the rate of cAMP hydrolysis to half of that of the vehicle-treated control reactions.
TNFα assays
Myelomonocytic U937 cultures growing in fresh medium (RPMI 1640 + 10% fetal bovine serum) at 106 cells/mL were preincubated for 17 h with 300 ng/mL PMA phorbol-12-myristate-13-acetate (PMA). Aliquots (105 cells) were diluted in equal volume of fresh medium and treated with compounds (10 µM final concentration) for 1 h in 96-well plates. Control cultures received vehicle only (ethanol containing <0.1% DMSO). Cultures were activated with 25 µg/mL LPS to stimulate TNFα release, 20 and culture supernatants were collected after 3 or 20 h of LPS stimulation. TNFα was measured using a human TNF enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer’s instructions. Statistical significance was determined using the Excel 1-tailed Student t-test function, with the differences being considered significant at p < 0.05.
Results
Identification of PDE7 inhibitors using a yeast growth assay
To identify PDE7 inhibitors, we constructed a fission yeast strain whose only PDE activity comes from the human PDE7A gene and whose growth behavior reflects its intracellular cAMP level. Using homologous recombination, we replaced the fission yeast cgs2
+ PDE gene with PDE7A as described for other mammalian PDE genes.
14
The CHP1189 screening strain also contains fbp1-ura4 and fbp1-lacZ reporter genes,
11
a deletion of the pap1
+ transcriptional activator (to enhance compound sensitivity),
21
and a gpa2
+ deletion to reduce cAMP synthesis to confer 5FOA-sensitive growth.
22
After determining growth conditions that optimize the detection of PDE7A inhibitors, 48,176 compounds (most at 20 µM;
In comparing the structures of the 24 compounds that conferred the highest average OD600 values (>0.821), 2 related compound families were immediately obvious. Two compounds are the steroids androsterone acetate (Microsource 00107113) and canrenone (Microsource 01505248), whereas another 3 are podocarpanes, which possess the same arrangement of three 6-carbon rings as found in steroids (
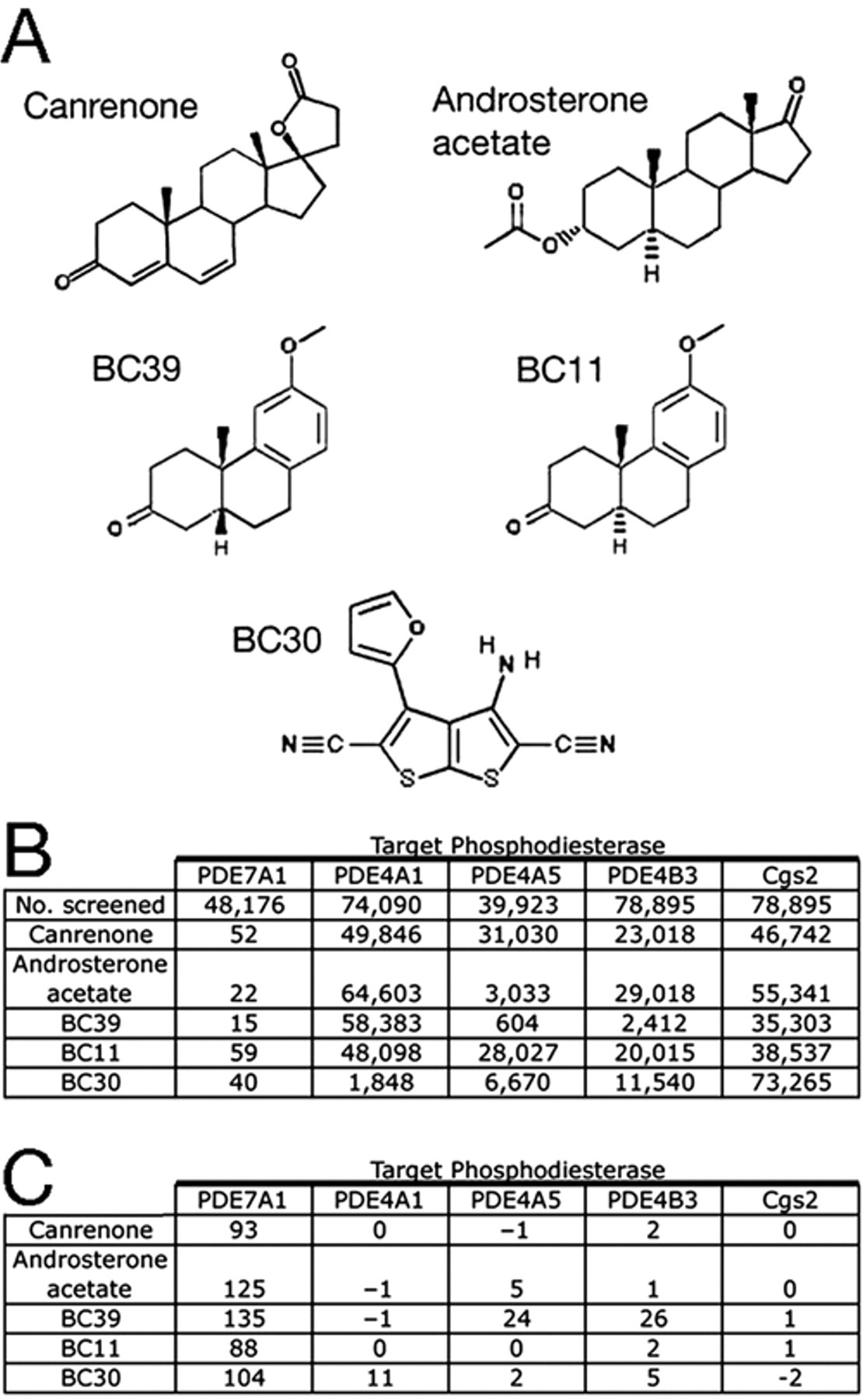
Structure and high-throughput screening (HTS) data for some PDE7A inhibitors. (
In addition to steroids and podocarpanes, an unusual heterocyclic compound was identified in this screen, 3-amino-4-(2-furyl) thieno[2,3-b]thiophene-2,5-dicarbonitrile (Maybridge KM03472SC), which we refer to as BC30 (
Characterization of PDE7A and PDE7B inhibition via yeast growth assays
As an initial test of compounds BC11, BC30, BC39, and BRL50481, we repeated the 5FOA growth assay on strains that express PDE7A or PDE7B, using a range of compound concentrations (
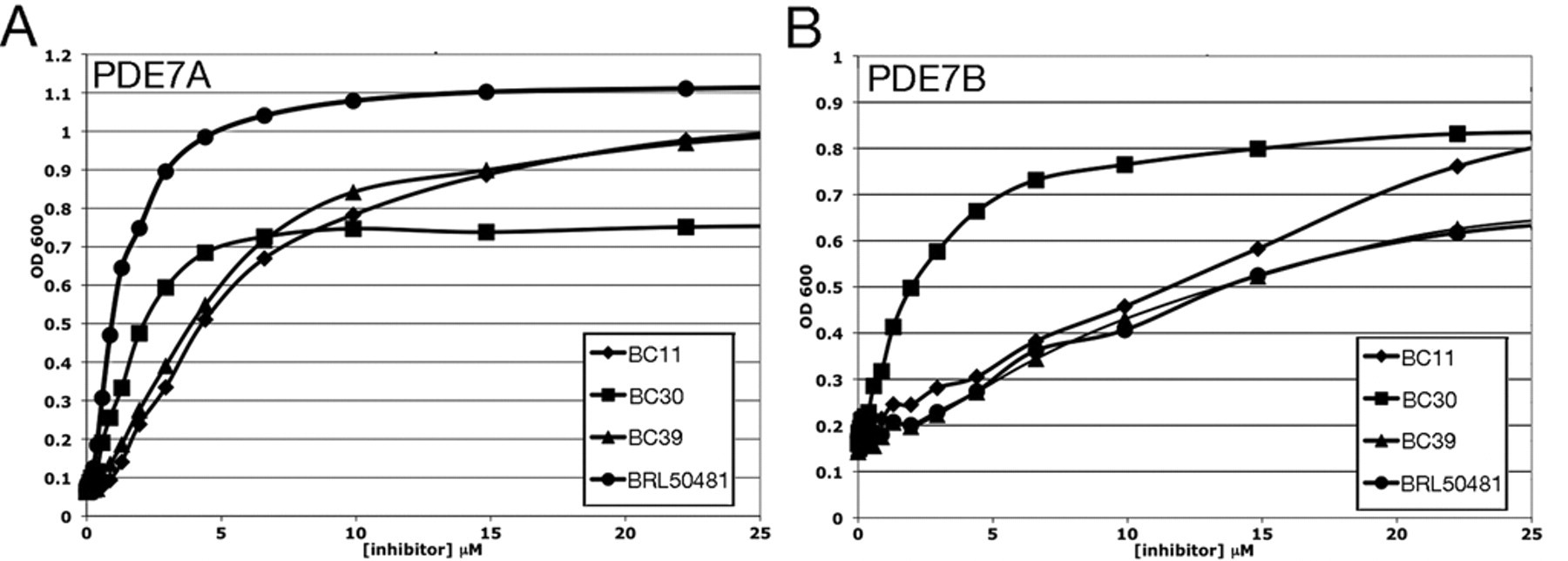
5FOA growth response curves of candidate PDE7A and PDE7B inhibitors. Optical density of cultures of a (
In vitro enzyme assays of PDE7A and PDE7B inhibition
Assays of purified PDE7A and PDE7B were carried out to compare the inhibitory potency of compounds BC11, BC30, BC39, and BRL50481 (
IC50 Values for PDE Inhibitors against PDE7A and PDE7B
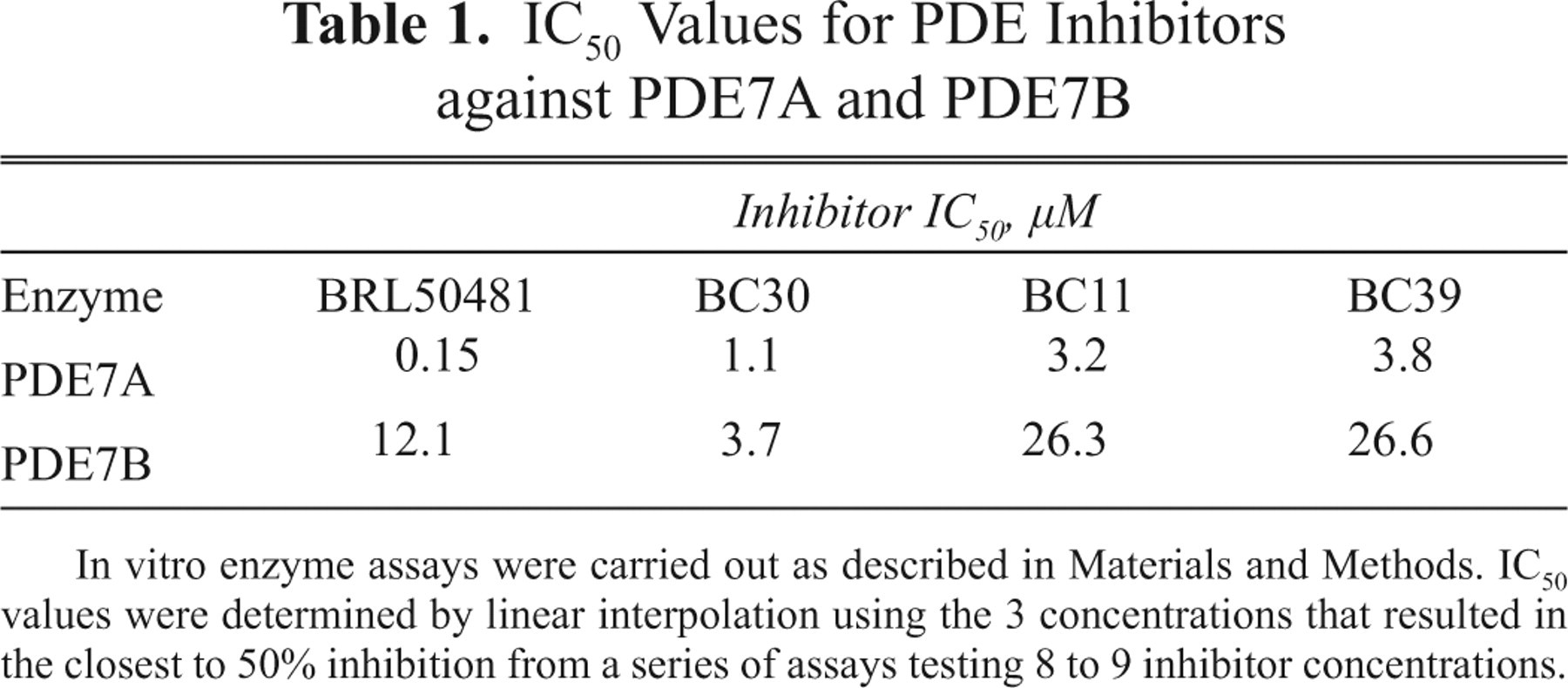
In vitro enzyme assays were carried out as described in Materials and Methods. IC50 values were determined by linear interpolation using the 3 concentrations that resulted in the closest to 50% inhibition from a series of assays testing 8 to 9 inhibitor concentrations.
BC30 activity against PDE4 in yeast-based and in vitro enzyme assays
Because PDE4 and PDE7 enzymes possess closely related catalytic domains and because BC30 weakly stimulates growth of a strain that expresses mouse PDE4A1 (
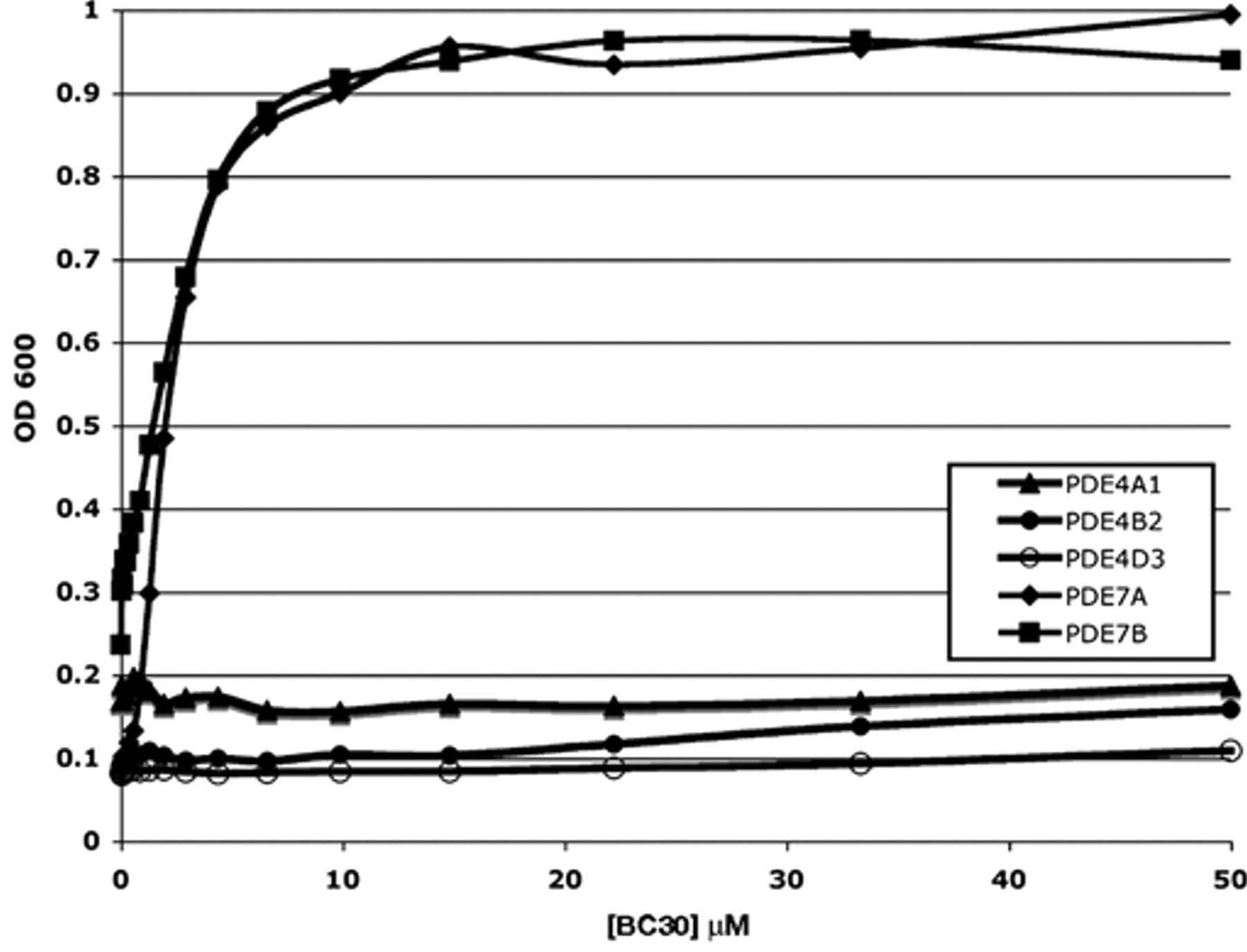
5FOA growth response curves for BC30 on strains expressing PDE4 or PDE7 enzymes. Assays were carried out as described in
BC30 was also examined as part of a panel of compounds in the in vitro enzyme assays using 2 µM compound and the catalytic domains from PDE4A, PDE4B, and PDE4D expressed in and purified from Escherichia coli.
19
Surprisingly, BC30 and 3 unrelated PDE7A inhibitors significantly stimulate cAMP hydrolysis by the PDE4D catalytic domain (residues 86-413 of PDE4D2; data not shown). BC30 acts at a remarkably low concentration; 25 nM produced the maximum increase in activity under conditions where the concentration of enzyme was 16 nM, doubling the Vmax of the enzyme (
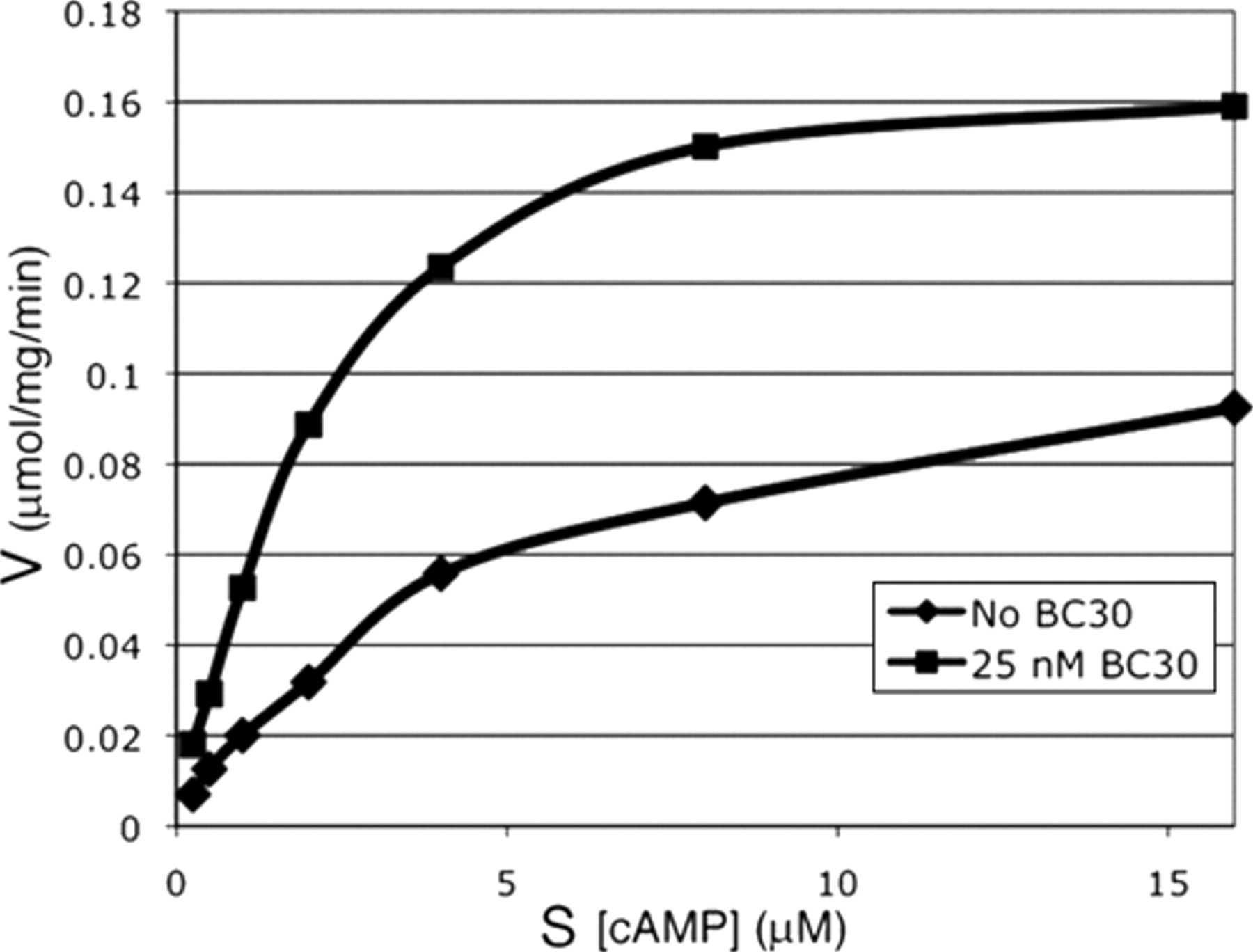
BC30 stimulation of cAMP hydrolysis by the PDE4D catalytic domain. In vitro enzyme assays were carried out using the PDE4D catalytic domain (PDE4D2 residues 86-413) in the absence or presence of 25 nM BC30 at varying concentrations of cAMP.
TNFα release assays assess anti-inflammatory potential of PDE7 inhibitors
BRL50481 was previously shown to enhance the anti-inflammatory effect of PDE4 inhibitors,
7
consistent with studies showing that PDE7A and PDE7B are significantly expressed in cells of the immune system.
27,28
We therefore examined the effect of BRL50481, BC11, BC30, and BC39 on TNFα release by LPS-treated U937 cells, alone or in combination with the PDE4 inhibitor rolipram. In vehicle-treated samples, we observed a nearly 50-fold increase in TNFα release as a result of LPS treatment after 3 h and a nearly 100-fold increase after 20 h (data not shown). Of the 5 compounds tested, only BC30 at both time points and rolipram at the 20-h time point conferred a significant reduction in TNFα release (
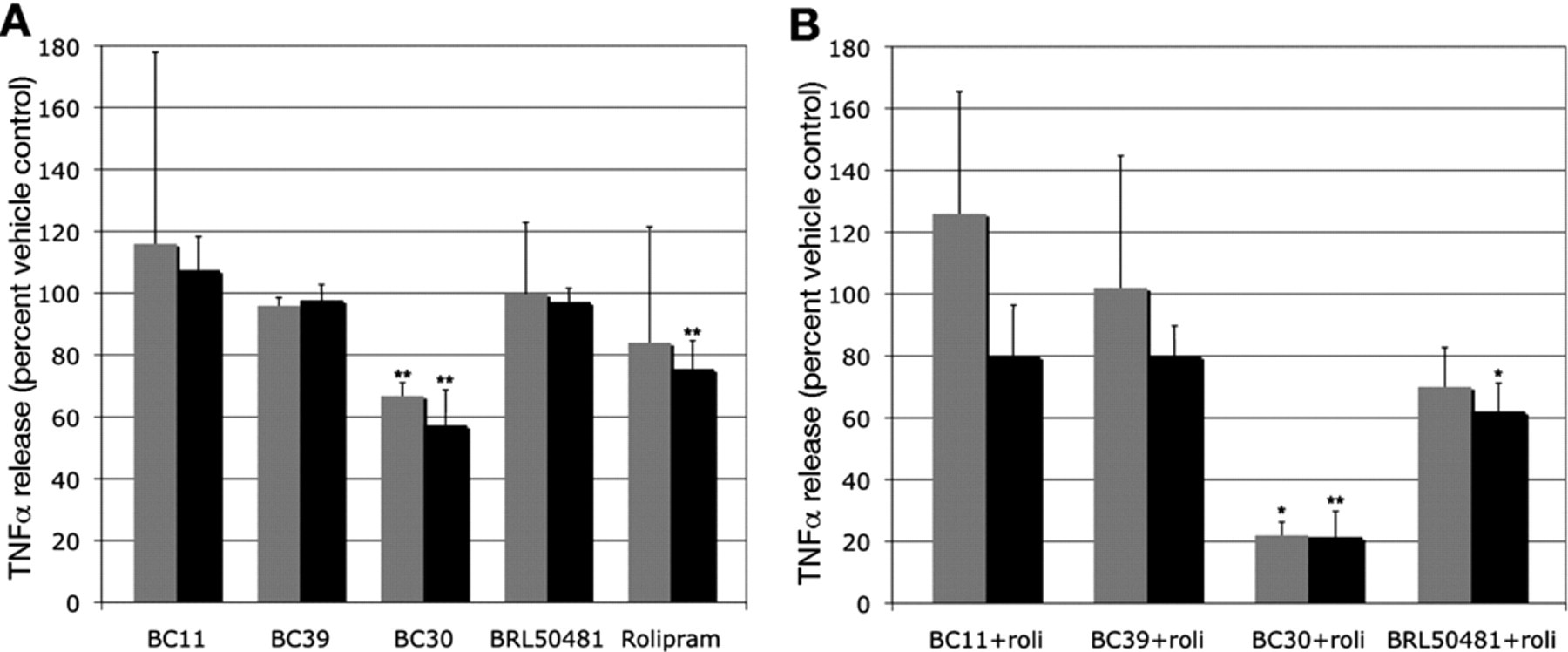
Effect of PDE inhibitors on lipopolysaccharide (LPS)-stimulated tumor necrosis factor α (TNFα) release in activated U937 cells. U937 cells were activated by PMA and then treated with LPS to stimulate TNFα production and release. The effect of a 1-h treatment with (
Discussion
The work presented here demonstrates that we have developed an effective fission yeast-based HTS for PDE7 inhibitors. Among the most potent inhibitors identified from over 48,000 compounds screened were 2 steroids and 3 podocarpanes, which are structurally related to steroids. This is noteworthy as the ICOS Corporation (Bothell, WA) previously identified a steroid-like compound, IC242, as a PDE7-specific inhibitor 23 ; however, as with most compounds under development by pharmaceutical companies, this compound is not readily available, nor has its structure been revealed. As BRL50481 is the only commercially available PDE7 inhibitor, we set out to compare it to some compounds identified in our screen, all of which are commercially available from either Microsource (Gaylordsville, CT) or Maybridge (Cornwall, UK).
BRL50481 was developed based on its ability to inhibit PDE7A as judged by in vitro enzyme assays, and in both the 5FOA cell growth assay and in vitro enzyme assays of PDE7A inhibition, it is superior to our PDE7 inhibitors. In contrast, BRL50481 is not nearly as effective for inhibition of PDE7B as judged by our cell-based assay and by in vitro enzyme assays (
An unusual observation from the in vitro enzyme assays is that several compounds originally identified as inhibitors of PDE7, including BC30, stimulate the activity of the PDE4D catalytic domain (
Finally, we show that BC30 reduces the inflammatory effect of LPS on activated U937 cells, as judged by TNFα release assays. In particular, we observe a potent synergistic effect on treatment with BC30 and rolipram (
The yeast growth-based screening method used to discover BC30 as a PDE7A/PDE7B inhibitor with potent anti-inflammatory properties requires that the compounds identified have drug-like characteristics in addition to being effective PDE inhibitors. These compounds must show little toxicity to fission yeast, which should exclude compounds that bind a large number of proteins, as such promiscuous binding would likely reduce cell growth. In addition, these compounds must be cell permeable to fission yeast and remain active for most or all of the 48-h incubation period of the assay. Therefore, compounds identified by this method may represent superior starting material with regard to drug development relative to compounds identified by in vitro enzyme assays that prioritize candidates solely on the basis of binding affinity. (At the same time, the relative potencies of the compounds discovered by this method are confirmed by in vitro enzyme assays [
Footnotes
Acknowledgements
We thank Joe Beavo and Miles Houslay for clones and for advice and encouragement during the course of this project; Nicola Tolliday, Lynn VerPlank, Jason Burbank, and Stephanie Norton for guidance on HTS; and Chip Stewart for assistance with statistical analyses. The project has been funded in part with federal funds from the National Cancer Institute’s Initiative for Chemical Genetics, National Institutes of Health, under contract no. N01-CO-12400, and has been performed with the assistance of the Chemical Biology Platform of the Broad Institute of Harvard and MIT.
This work was supported by a grant from Boston College and by NIH grant R21 GM079662 to CSH.