
Abstract
The development of multidrug resistance (MDR) remains a significant obstacle in treating cancer patients with chemotherapy. To identify small-molecule compounds that can reverse MDR, the authors used a cell-based screening assay with an MDR ovarian cancer cell line. Incubating MDR cells with a sublethal concentration of paclitaxel in combination with each of 2000 small-molecule compounds from the National Cancer Institute Diversity Set Library, they identified NSC77037. The cytotoxic activity of NSC77037 and the duration of its effect were evaluated in vitro using a panel of cancer cell lines expressing permeability glycoprotein (Pgp), multiple drug resistance protein 1 (MRP 1), and breast cancer resistance protein (BCRP). The mechanism of its effects was further analyzed by assessing the retention of calcein and Pgp-ATPase activity. The relative potency of MDR reversal by NSC77037 was significantly higher than that of frequently used MDR reversal agents such as verapamil and cyclosporine A. NSC77037 reversed Pgp without reversing MRP or BCRP-mediated MDR. NSC77037, at a concentration of >10 µM, moderately inhibited the proliferation of both sensitive and resistant cell lines, but the inhibitory effect of NSC77037 was not altered by coincubation with the Pgp inhibitor verapamil, suggesting that NSC77037 itself is not a substrate of Pgp. NSC77037 directly inhibited the function of Pgp in a dose-dependent manner, but it did not alter the protein expression level of Pgp. The use of NSC77037 to restore sensitivity to chemotherapy or to prevent resistance could be a potential treatment strategy for cancer patients.
Introduction
M
The MDR1 (ABCB1) gene product Pgp is a member of the ABC (ATP binding cassette) superfamily of transporter proteins that acts as an energy-dependent drug efflux pump, preventing intracellular accumulation of a variety of cytotoxic drugs. Intrinsic or acquired expression of Pgp is a major problem in cancer chemotherapy. 2,4 Pgp expression has been linked to chemoresistance and decrease in survival in a variety of cancers. 1,2,5 Overexpression of the ABCB1 gene within drug-sensitive cells or mice to produce transgenic animals confers resistance to such agents as anthracyclines, paclitaxel, and the Vinca alkaloids. 6,7 Recurrent tumors and tumors that progress after chemotherapy often have a population of cells that have developed MDR, and eradication of such cells during initial treatment or at the time of recurrence is crucial in achieving greater efficacy from chemotherapy.
The development and discovery of agents that reverse MDR phenotype with high efficiency and low toxicity have been the focus of extensive research, but previously reported compounds have been nonspecific and weak in potency. 3,8 Among these compounds, clinical toxicities associated with their use at the required concentrations to inhibit Pgp function have prohibited their widespread utility. 3 First-generation Pgp inhibitors such as verapamil had lower affinity to Pgp than to other target proteins (e.g., Ca2+ channel) and thus when administered at sufficiently high doses caused cardiac toxicity. 9 Second-generation inhibitors have been reported to possess the potential to change the pharmacokinetics of conventional chemotherapeutic drugs by altering their metabolism and clearance from the body, presumably through interactions with cytochrome P450. 10 These inhibitors competed with the chemotherapeutic drugs for metabolism as well as for efflux. 11 Results from phase III clinical trials using third-generation Pgp inhibitors have been disappointing, and to date, no survival benefits by any Pgp inhibitor have yet been achieved. 3,8,12
The current methods commonly used to elucidate complex biologic systems have their limitations and strengths, but screening of a large compound library has the potential to overcome these obstacles. It has been shown that screening of chemical libraries for biologically active compounds aimed at certain cellular targets is unbiased, reproducible, and efficient. 13-16 To overcome MDR and improve cancer treatments, we have undertaken a rational design for the development, optimization, and validation of an MDR cell-based screening assay using 2000 small-molecule compounds from the National Cancer Institute (NCI) Diversity Set Library. NSC77037, identified through this novel screening assay as one of the most potent MDR reversal agent, holds promise for the treatment of variety of MDR cancers.
Materials and Methods
Cell lines and antibodies
The paclitaxel-resistant SKOV-3TR, OVCAR8TR, U-20STR, MCF-7TR, and SW480TR lines as well as doxorubicin-resistant MCF-7DR were established as previously reported. 17-20 The human ovarian cancer cell line SKOV-3, human osteosarcoma cell line U-20S, KHOS, human uterine sarcoma cell line MESSA and its doxorubicin-selected drug-resistant cell line MESSA/Dx5, human breast cancer cell line MCF-7, human colon cancer cell line SW480, and human non–small cell lung cancer cell line H-69 and its doxorubicin-selected drug-resistant cell line H-69AR were obtained from the American Type Tissue Collection (Rockville, MD). Dr. Patricia Donahoe (Massachusetts General Hospital, Boston, MA) provided the human OVCAR5 and OVCAR8 ovarian cancer cell lines. Dr. Efstathios Gonos (Institute of Biological Research & Biotechnology, Athens, Greece) provided the doxorubicin-resistant U-20S R2 (referred in the text below as U-20SDR) and KHOS R2 cell lines. Dr. Stephen B. Howell (University of California Medical Center, San Diego, CA) provided the cisplatin-resistant ovarian cancer 2008cp70 and IGROV1cp cell lines. Dr. Erasmus Schneider (Wadsworth Center, Albany, NY) provided the mitoxantrane-resistant breast cancer MCF-7/MX cell line. Dr. Katia Scotlandi (Institute Orthopedics Rizzoli, Italy) provided ET-743-resistant TC-ET 6nM and TC-ET 12nM cell lines.
The Pgp monoclonal antibody C219 was purchased from Signet (Dedham, MA). The mouse monoclonal antibody to multiple drug resistance protein 1 (MRP1) and 3-(4, 5-dimethylthyazol-2-yl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) reagent were purchased from Sigma-Aldrich (St. Louis, MO). The monoclonal antibody to the breast cancer resistance protein (BCRP) antibody was purchased from Chemicon (Temecula, CA). The goat antirabbit–horseradish peroxidase (HRP) and goat antimouse-HRP were purchased from Bio-Rad (Hercules, CA). SuperSignal® West Pico Chemiluminescent Substrate was purchased from PIERCE (Rockford, IL).
Cell culture
The cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 µg/mL streptomycin (Invitrogen, Carlsbad, CA). Cells were incubated at 37°C in 5% CO2–95% air atmosphere and passaged when near confluent monolayers were achieved using trypsin-EDTA solution. Drug-resistant cell lines were periodically cultured in the respective drug to confirm their drug resistance characteristics. Cells were free from mycoplasma contamination as tested by the MycoAlert® Mycoplasma Detection Kit from Cambrex (Rockland, ME).
Drugs and chemicals
The Structural Diversity Set is a library comprising approximately 2000 small molecules derived from more than 140,000 compounds available on plates through the NCI. Verapamil and DMSO were purchased from Sigma-Aldrich. Paclitaxel, doxorubicin, and cisplatin were obtained through unused residual clinical material provided by the pharmacy at the Massachusetts General Hospital. ET-743 and PM00104 were supplied by PharmaMar (Spain). The stock solution of drugs was prepared according to the drug specifications and stored at –20°C.
Cell-based drug screening assay
We have previously described methods for this assay. 16 Briefly, SKOV-3TR cells were treated with paclitaxel for initial screening. SKOV-3TR showed 100-fold higher resistance to paclitaxel as compared with parental sensitive cells and was able to grow in a 0.3-µM concentration of paclitaxel. Screening conditions using SKOV-3TR were optimized in 96-well plates for growth conditions, small molecular compound and drug concentrations, and assay times prior to screening. The experiments were conducted in 2 control plates (A and B) and 1 experimental plate (C) to permit plate-to-plate comparisons. On day 1, SKOV-3TR cells were seeded at 2 × 104 cells/well on 96-well plates labeled A, B, and C and were incubated for 24 h at 37°C. On day 2, 0.1 µM paclitaxel was added to plate A, 1 µM of different small molecular compounds without paclitaxel was added to plate B, and mixtures of both small molecular compound and 0.1 µM paclitaxel were added to plate C. From days 4 to 6, fresh medium was replaced when necessary as described above and evaluated for cytotoxicity under the microscope. The control plates (A and B) were used to evaluate cytotoxicity of paclitaxel and the small molecular compound itself and to exclude compounds that were lethal to the cells in the absence of chemotherapeutic drug. Plate C was given both small molecular compound and 0.1 µM paclitaxel. This concentration is a sublethal dose of paclitaxel for the SKOV-3TR cell line, and a positive result would be cell death at 4 to 6 days. On day 6, the number of viable cells was evaluated for growth inhibition under the microscope manually, and final results were determined via CellTiter 96®AQueous One Solution Cell Cytotoxicity Assay (Promega, Madison, WI) by a SPECTRAmax® Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA). Small molecular compounds that were associated with cell survival in plates A and B and death in plate C were identified as “hits” and selected for further study. Once hits were identified, the results were validated in other drug-resistant cell lines.
In vitro cytotoxicity assay
Drug cytotoxicity was assessed using the MTT assay as previously described. 21 Briefly, 2 × 103 cells per well were plated in 96-well plates in culture medium containing increasing concentrations of drug. After 7 days of culture, 10 µL MTT (5 mg/mL in phosphate-buffered saline [PBS]) was added to each well, and the plates were incubated for 4 h. The resulting formazan product was dissolved with acid-isopropanol, and the absorbance at a wavelength of 490 nm (A490) was read on a SPECTRAmax® Microplate Spectrophotometer (Molecular Devices). The absorbance values were normalized by assigning the value of the control line in the medium without drug to 1.0 and the value of the no-cell control to 0. Experiments were performed in triplicate.
Duration of MDR reversal
The effect of the compounds on the duration of MDR reversal was assessed as previously described. 22 In brief, 8 × 105 SKOV-3TR cells/mL were incubated for 24 h with NSC77037 or verapamil before being washed 3 times with the medium. The cells were then incubated for 4 days with the addition of varying concentrations of paclitaxel. Final results were determined by MTT assay.
Western blot analysis
Pgp, MRP1, and BCRP proteins were analyzed in total cell lysates. Total cell lysates were prepared, and Western blot analysis was performed. Briefly, the cells were lysed in 1 × RIPA lysis buffer (Upstate Biotechnology, Charlottesville, VA), and protein concentration was determined by the DC Protein Assay (Bio-Rad). Then, 25 µg of total protein was resolved on NuPage™ 4% to 12% Bis-Tris Gels (Invitrogen) and immunoblotted with specific antibodies. Primary antibodies were incubated in Tris-buffered saline (TBS; pH 7.4) with 0.1% Tween-20 with gentle agitation overnight at 4°C. HRP-conjugated secondary antibodies (Bio-Rad) were incubated in TBS (pH 7.4) with 5% nonfat milk (Bio-Rad) and 0.1% Tween-20 at a 1:2000 dilution for 1 h with gentle agitation at room temperature. Positive immunoreactions were detected by using SuperSingal® West Pico Chemiluminescent Substrate.
Drug efflux assay
The Vybrant™ multi–drug resistance assay kit (Molecular Probes, Eugene, OR) was used to measure the drug efflux properties of different resistant cell lines as previously described. 23 Drug-sensitive and drug-resistant cells (50,000 cells/well) were cultured in a 96-well plate overnight. Triplicated cultures of cells were treated with NSC77037 or verapamil for 1 h and then incubated in calcein AM in 100 µL total volume. After 30 min, the cells were washed and centrifuged twice with 200 µL cold RPMI 1640 culture medium, and cell fluorescence was measured at a wavelength of 490 nm (A490) on a SPECTRAmax® Microplate Spectrophotometer (Molecular Devices).
Fluorescence microscopy
For visualization of the effects of NSC77037 on the intracellular retention of calcein, 10,000 resistant cells were seeded onto Lab-Tek 8-well chamber slides on the day prior to the assay. Cells were then incubated with either 0.25 µM calcein AM alone or in the presence of NSC77037 in RPMI 1640 for 1 h at 37°C. Images were acquired by a Nikon Eclipse Ti-U fluorescence microscope (Nikon Corp., Tokyo, Japan) equipped with SPOT RT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI).
PGP-ATPase assay
The Pgp-Glo™ Assay Systems (Promega) was used to perform luminescent Pgp-ATPase assays. 24 This assay provides a rapid, colorimetric, compound-independent measure of the concentration dependence of any interaction of a drug with Pgp. If a drug does not stimulate Pgp-ATPase activity, the assay can still determine if the drug interacts with Pgp as an inhibitor of the ATPase activity stimulated by a known substrate, such as verapamil. The effect of NSC77037 on the ATPase activity of Pgp was measured according to the manufacturer’s protocol. Luminescence was read on a BMG LABTECH Polarstar Optima Luminometer (Cary, NC).
Statistical analysis
Student’s t-test was used to compare the differences between groups (GraphPad PRISM® 4 software, GraphPad Software, San Diego, CA). Results are given as mean ± SD, and results with p < 0.05 were considered statistically significant.
Results
NSC77037 reverses MDR
To identify candidates of small molecular compounds that can reverse drug resistance, we screened the NCI Diversity Set Library for small-molecule compounds that reconstitute paclitaxel cytotoxicity in a Pgp-expressing, MDR human ovarian cancer cell line SKOV-3TR. We determined that 0.1 µM paclitaxel is the concentration of paclitaxel that is lethal to drug-sensitive SKOV-3 cells but not to SKOV-3TR cells. When combined with 0.1 µM paclitaxel, 1 µM NSC77037 induced significant cell death in SKOV-3TR, which grows well in either 0.1 µM paclitaxel or 1 µM NSC77037 ( Fig. 1A ). After screening more than 2000 small molecular compounds in the NCI Diversity Set Library, NSC77037 and NSC23925, 2 small-molecule inhibitors, were identified as agents with the highest activities ( Fig. 1B ). We have described the results of NSC23925 in a prior report. 16
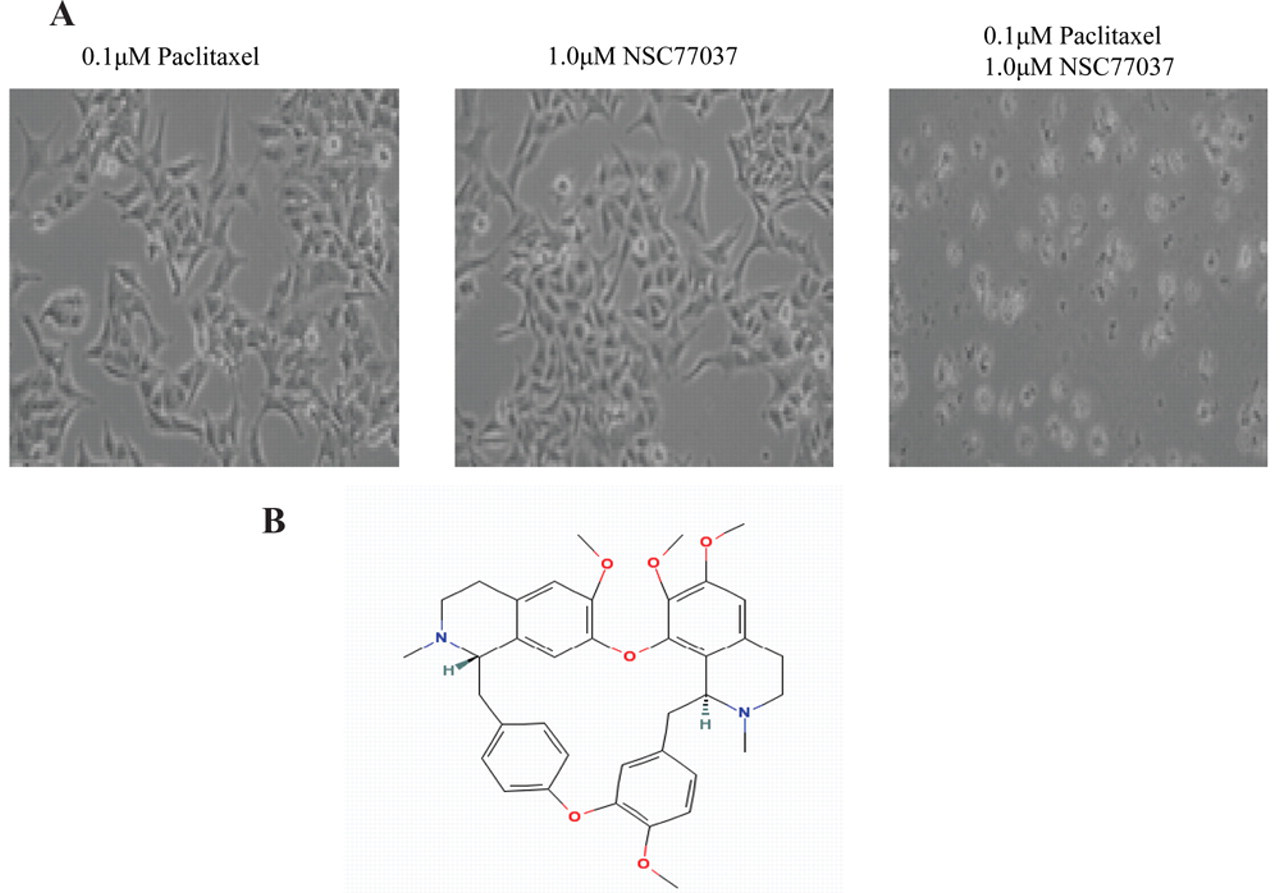
Reversal of paclitaxel resistance by NSC77037 and its chemical structure. (
Reversal of drug resistance in a panel of MDR cell lines
To determine whether NSC77037 is able to reverse drug resistance in human cancer cell lines, we evaluated several MDR cell lines with a variety of anticancer agents. We tested for a wide variety of tumor types to show that NSC77037 was able to reverse resistance to all MDR cell lines in which Pgp is highly expressed, including ovarian cancer MDR cell lines SKOV-3TR and OVCAR8TR, breast cancer MDR cell line MCF-7TR, and sarcoma MDR cell lines MESSA/Dx5, KHOS R2, and U-20SDR (representative data from SKOV-3TR, OVCAR8TR, and KHOS R2 shown in Table 1 ). The results are from 3 independent experiments with triplicate wells. Maximal reversal of MDR was typically seen with NSC77037 in doses around 1 µM ( Fig. 2 ). There was no significant change in reversal of MDR after 1 µM (data not shown). NSC77037 was highly active across the panel of cell lines and demonstrated significant reversal of resistance when used in conjunction with paclitaxel, docetaxel, doxorubicin, daunorubicin, gemcitabine, vincristine, ET-743, or PM00104. The potency of NSC77037 was 10- to 50-fold greater than that of verapamil or CsA. Moreover, the presence of <5 to 8 µM of NSC77037 alone had no cytotoxic effect in the parental cell lines SKOV-3 and OVCAR8. Importantly, NSC77037 did not alter the cytotoxicity of cisplatin and methotrexate, both agents known to be unaffected by MDR1 mechanisms (see Supplemental Table 1 at http://jbx.sagepub.com/supplemental). These results suggest that reversal of resistance was mostly attributable to the inhibition of Pgp. Furthermore, we tested other cell lines with non-Pgp-mediated MDR mechanisms (H-69/AR cells that express MRP1, but not Pgp, and MCF-7 MX cells that express BCRP, but not Pgp) to evaluate the specificity of NSC77037. We observed that NSC77037 was unable to reverse drug resistance in these non-Pgp-expressing drug-resistant cell lines, suggesting that NSC77037 is a specific inhibitor of Pgp.
Effect of NSC77037 Reversing Drug Resistance in Multidrug Resistant Cell Lines
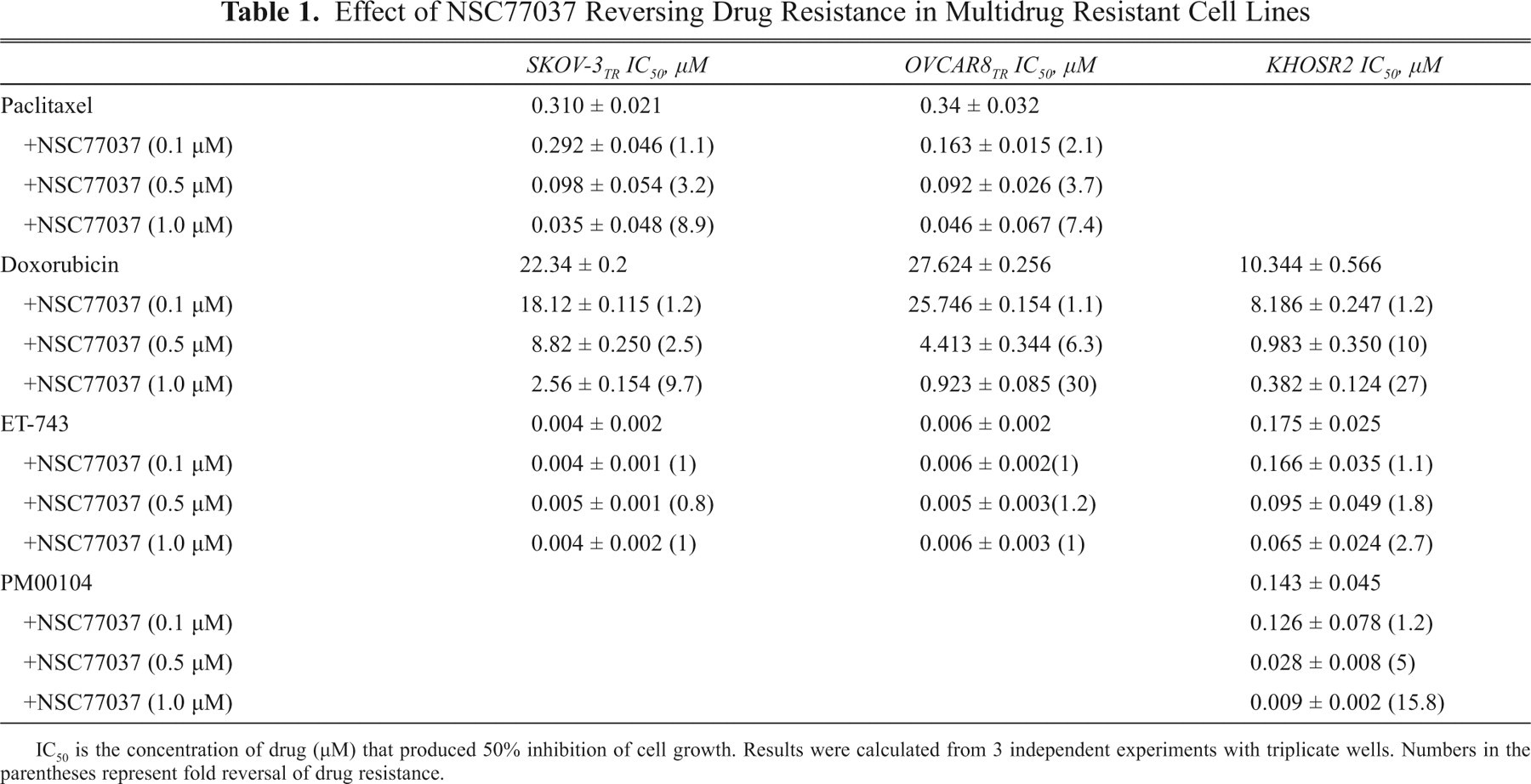
IC50 is the concentration of drug (µM) that produced 50% inhibition of cell growth. Results were calculated from 3 independent experiments with triplicate wells. Numbers in the parentheses represent fold reversal of drug resistance.
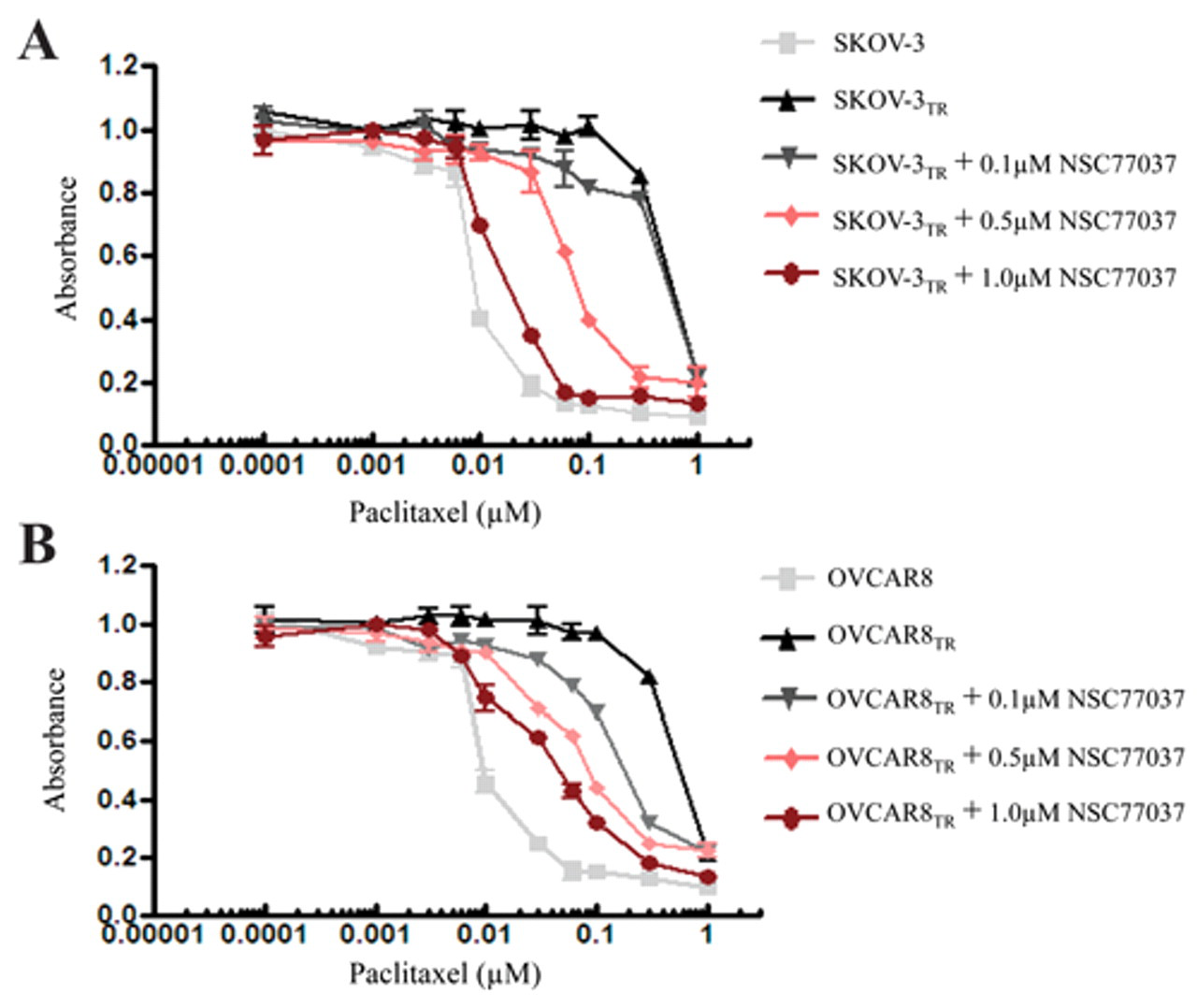
Effect of NSC77037 on reversing drug resistance in MDR cell lines. (
NSC77037 is not a Pgp substrate
Although NSC77037 reversed Pgp-mediated MDR in the nanomolar concentration range, NSC77037 was not cytotoxic by itself at doses up to 10 µM in both sensitive and resistant cell lines. At high concentrations, NSC77037 by itself is equally inhibitory on the proliferation of both SKOV-3/SKOV-3TR and OVCAR8/OVCAR8TR cell lines in a dose-dependent manner ( Fig. 3A ). IC50s were similar in matched cell lines that did and did not express Pgp. The IC50 for NSC77037 was 7 µM in SKOV-3/SKOV-3TR and 20 µM in OVCAR8/OVCAR8TR cell lines ( Fig. 3A ), whereas the mean concentration of NSC77037 required for maximal reversal of resistance in SKOV-3TR or OVCAR8TR to various cytotoxic drugs was 0.5 to 1 µM ( Fig. 2A,B ). Thus, NSC77037 begins to show toxicity to human cell lines only at concentrations 14- to 40-fold higher than those required for maximal reversal of drug resistance. These data indicate that overexpression of Pgp does not confer resistance to NSC77037. As described in other studies, these results suggest that a compound such as NSC77037 is not a Pgp transport substrate. 22,25,26 To validate this, we assessed the effect of the Pgp inhibitor, verapamil, on NSC77037 sensitivity in Pgp-overexpressing SKOV-3TR cells. As expected, verapamil did not influence the inhibitory effect of NSC77037 in SKOV-3TR cells ( Fig. 4A ). Similar results were found in OVCAR8TR (data not shown). These data collectively demonstrate that NSC77037 itself is not a substrate of Pgp, and Pgp does not confer resistance to NSC77037 in cancer cells. Also, NSC77037 had no cross-resistance to other cytotoxic agents such as paclitaxel and doxorubicin. These results are of clinical significance because they suggest that NSC77037 may be a good option for the patients with tumors that have already developed MDR.
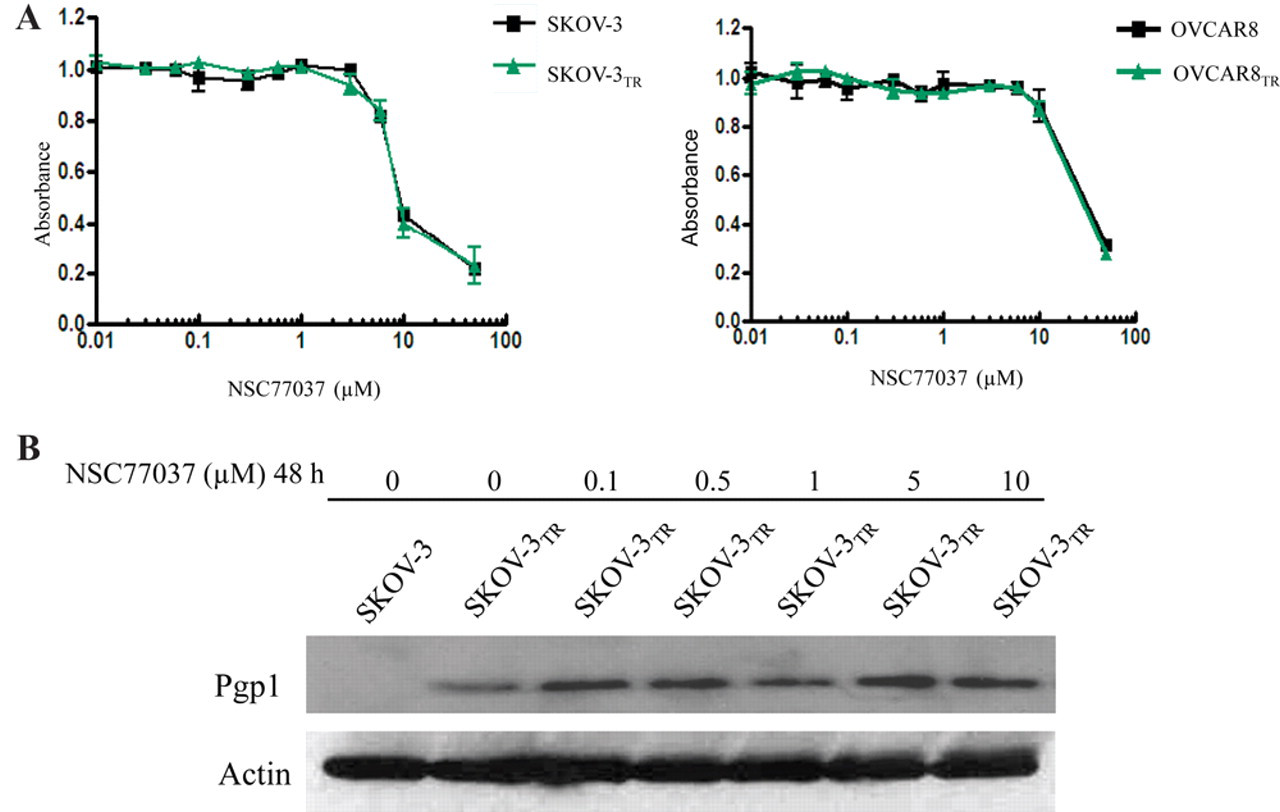
Nonspecific toxicity of NSC77037 and effect of NSC77037 on expression of Pgp. (
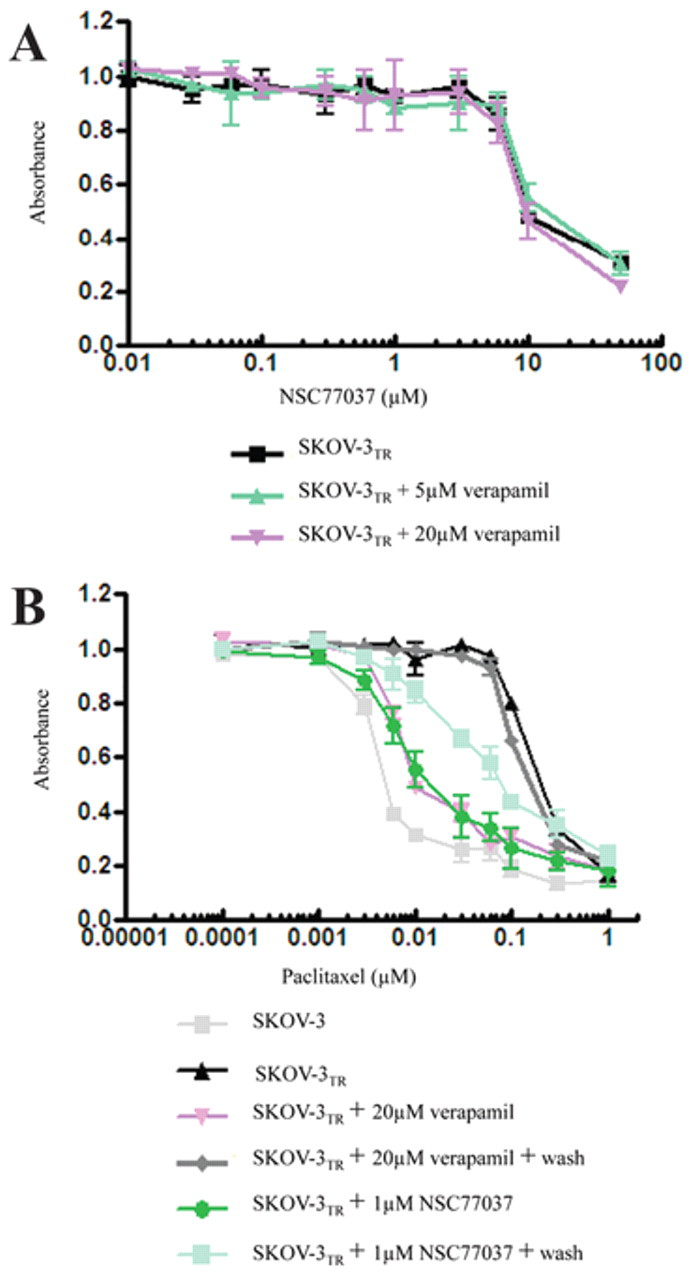
Effect of verapamil on NSC77037 sensitivity in Pgp-overexpressing cells and duration of NSC77037 activity. (
Duration of NSC77037 activity
We observed that NSC77037 inhibits the efflux of both paclitaxel and doxorubicin from the MDR cells lines SKOV-3TR and OVCAR8TR, and we also observed that NSC77037 remains effective even after it was washed out from the efflux medium subsequent to overnight exposure (1 µM; Fig. 4B ). NSC77037 was effective at inhibiting Pgp-mediated transport for at least 4 days after its washout from the medium. In comparison, verapamil, even with a very high concentration (20 µM), displayed only a short duration (24 h) of inhibition ( Fig. 4B ). Similar results were obtained in OVCAR8TR cells (data not shown).
NSC77037 modulates Pgp-mediated uptake and efflux of calcein AM
NSC77037 was shown to increase intracellular accumulation of calcein in MDR cell lines in a dose-dependent manner as determined through both image analysis ( Fig. 5A,B ) and microplate spectrofluorometer analysis ( Fig. 5C,D ). NSC77037 had a prominent effect on the accumulation of calcein in these cells at a concentration as low as 1 nM. Half-maximal reversal of accumulation deficit was observed at 100 nM and near maximal at 600 nM. The potency of NSC77037 was significantly greater than verapamil or CsA ( Fig. 5 ). In the control parental drug-sensitive cell lines SKOV-3 and OVCAR8, which do not overexpress Pgp, NSC77037 had no evident effect on accumulation of calcein ( Fig. 5A,C ).
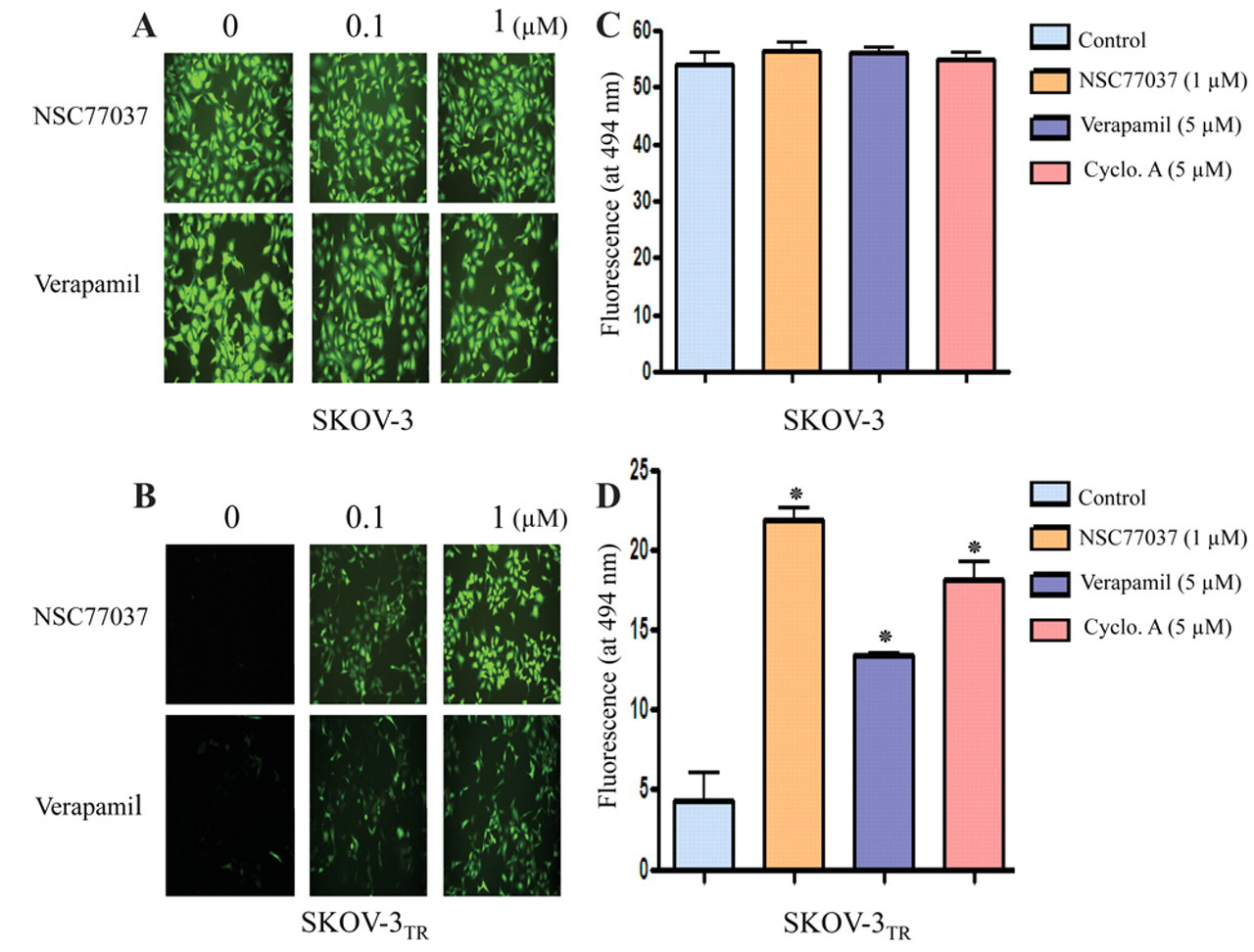
Calcein AM efflux from SKOV-3 and SKOV-3TR cells. The calcein AM assay was optimized and performed using the Vybrant Multidrug Resistance kit. Cells were seeded at 50,000 cells/well in a 96-well plate and incubated for 24 h. Cells were treated with either NSC77037 or verapamil for 1 h and then incubated with calcein AM for 30 min. The cell fluorescence images were acquired by a fluorescence microscope (
NSC77037 stimulates Pgp ATPase activity, but not Pgp expression
The increased accumulation of drug in MDR cells may be due to decreased expression or function of Pgp. 16 We monitored ATPase activity in cell membrane preparations as a method for identifying compounds that interact with the drug efflux transporters. Pgp exhibits a highly drug-dependent adenosine triphosphate (ATP) hydrolysis activity, and a variety of Pgp inhibitors and substrates can stimulate ATPase activity. 25-28 In this assay, a test molecule is considered to interact specifically with Pgp if it significantly modulates ATPase activity, at any concentrations from 0.05 to 50 µM. Using this method, we examined the effect of NSC77037 on both the expression and function of Pgp. NSC77037 significantly increased the ATPase activity in purified recombinant human Pgp membrane protein in a dose-dependent manner ( Fig. 6 ). However, the expression level of Pgp was not affected by different concentrations of NSC77037 (up to 10 µM for 48 h; Fig. 3B ). Similar to our findings on NSC23925, these results suggest that NSC77037 stimulates transporter ATPase activity by directly interacting with Pgp. 16
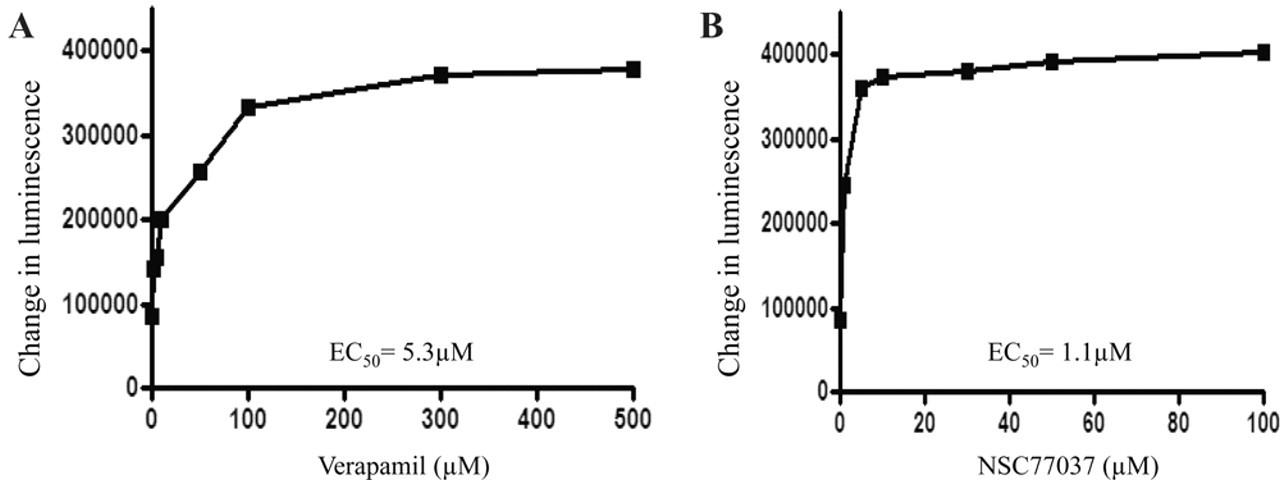
Stimulation of Pgp-ATPase activity by NSC77037 compared to verapamil. Untreated, Na3VO4, verapamil, and NSC77037-treated Pgp reactions were performed according to the manufacturer’s protocol. Basal Pgp-ATPase activity was calculated by subtracting luminescence of untreated samples from luminescence of Na3VO4. Change in luminescence between the test compounds and basal Pgp-ATPase activity was plotted against test compound concentrations. (
Discussion
In this study, a cell-based screening assay using the MDR cancer cell line was used to screen the NCI Structural Diversity Set and identify small-molecule compounds capable of reversing MDR. Cell-based screening technique has the advantage of unbiased testing of a large number of small-molecule compounds that could potentially reverse drug resistance. The results demonstrate that the assay is sensitive, reproducible, and robust.
One of the most potent small molecular compounds in our screen that was capable of reversing the MDR activity was NSC77037/tetrandrine, an agent that has been described by several groups in China to reverse MDR in Pgp overexpressing cell lines. 29,30 We add to this body of literature by further characterizing tetrandrine activity and the mechanism in a panel of human cancer cell lines resistant to a variety of chemotherapeutic agents. We observed that tetrandrine can reverse MDR in various cell lines. We also observed that tetrandrine reverses resistance to a panel of anticancer agents such as paclitaxel, doxorubicin, and ET-743.
Tetrandrine, a bis-benzylisoquinoline alkaloid compound, was isolated from the root of Stephania tetrandra and is used in traditional Chinese medicine as an antirheumatic, anti-inflammatory, and antihypertensive agent. 31 Some studies have shown that tetrandrine possesses antitumor activity in cultured tumor cells and animal models. 32,33 Tetrandrine induces G1-phase arrest in cells containing wild-type p53 and apoptosis in both p53 wild-type and null cells. 33 More recently, studies showed that the tetrandrine-derived compound, bromotetrandrine, can reverse doxorubicin resistance in the breast cancer cell line both in vitro and in vivo. 29,34 Tetrandrine has also been used in combination with daunorubicin, etoposide, and cytarabine (TET-DEC) in treating acute myelogenous leukemia (AML) patients with poor prognosis. 35 TET-DEC was relatively well tolerated in these AML patients with poor risk and had encouraging antileukemic effects.
Tetrandrine has been reported to reverse drug resistance previously, but in this study, we further showed that tetrandrine stimulates ATPase activity in a dose-dependent manner, which is required for the proper function of Pgp. 36 Although ATPase activity is closely associated with the function of Pgp, an increase in ATPase activity does not necessarily lead to an enhancement of Pgp function. Verapamil, which is a Pgp inhibitor that stimulates the ATPase activity of Pgp, also inhibits Pgp function. 26 MDR inhibitor, 5, 7, 30, 40, 50-pentamethoxyflavone (PMF), stimulates the ATPase activity in a dose-dependent manner. 37 Gefitinib, an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, also stimulates ATPase activity of Pgp and directly inhibits the function of Pgp in MDR cancer cells. 38 We showed that tetrandrine inhibits the efflux of Pgp substrates such as paclitaxel and doxorubicin without affecting the expression level of Pgp, suggesting that tetrandrine is capable of reversing MDR in cancer cells by directly inhibiting its function rather than content.
We further elucidated the characteristics using several different assays. Full to partial reversal of resistance in several MDR cell lines to several major classes of chemotherapy drugs was achieved in the presence of 10 to 2000 nM tetrandrine. Direct comparison with several modulators such as verapamil and CsA in different assays demonstrated that tetrandrine is the most potent among the 3 modulators. Tetrandrine showed 20-fold more potency than verapamil and 50-fold more potency than CsA. In addition, these studies clearly indicated that reversal of MDR by tetrandrine was through selective and potent inhibition of Pgp function. In contrast to the modulatory activity in the resistant cell lines, tetrandrine had no significant effect on cytotoxic drug activity in non-Pgp-expressing parental cell lines, nor did it affect the cytotoxicity of non-Pgp substrates such as cisplatin and methotrexate. Further confirmation of selectivity of tetrandrine for Pgp was provided because tetrandrine, even at very high concentrations (50 µM), did not inhibit MRP1 or BCRP function. These findings provide strong support for further development of tetrandrine or its derivates as candidate inhibitors of MDR in cancer treatments.
Footnotes
Acknowledgements
This project was supported in part by a grant from the Gattegno and Wechsler funds. Dr. Duan is supported in part through a grant from Ovarian Cancer Research Foundation (OCRF), Sarcoma Foundation of America, and the National Cancer Institute, NIH (Nanotechnology Platform Partnership), R01-CA119617. Dr. Choy is supported by the Jennifer Hunter Yates Foundation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.