
Abstract
PEGylation modification has been used to improve the pharmacokinetic properties of protein-based drugs. For example, PEGylated human growth hormone (hGH) has been shown to exhibit better pharmacokinetic profiles than the unmodified hGH. Unlike chemical PEGylation of hGH that is difficult to be controlled to result in homogeneity, microbial transglutaminase (mTGase) only conjugates poly(ethelene glycol) (PEG) on glutamine-40 (Q40) and glutamine-141 (Q141) of hGH, the only glutamine residues exposed. Yet, an mTGase that can selectively conjugate PEG to only 1 glutamine residue is more desirable to control the homogeneity of the product. In this study, the authors have developed a novel high-throughput assay, with which they have identified 5 mTGase mutants that are highly specific for conjugating PEG to Q141 of hGH. In this scintillation proximity assay (SPA)–based method, the authors have (1) achieved a high expression level of active mTGase, which is toxic to the living cell, directly from Escherichia coli (0.2 U/mL/OD600) by in vivo activation; (2) developed a high-throughput affinity purification method to eliminate the strong interference of cellular protein to mTGase reaction; and (3) used therapeutic protein as the substrate. This method is highly sensitive, is easily automated, and could be generally applied to screening mTGases with desired specificity targeting on different therapeutic proteins.
Introduction
PEG
Among the chemical and enzymatic strategies, site-specific PEGylation mediated by microbial transglutaminases (mTGase) has been investigated for pharmaceutical application. 9-12 TGases, also known as protein-glutamine γ-glutamyltransferase (E.C.2.3.2.13), catalyze the acyl-transfer reactions between the γ-carboxyamide group of the glutamine residues of a protein/peptide and the free amine group. 13 They show catalytic activity on selected glutamine residues that are exposed on the surface of native protein. For example, our previous data showed that among the 13 glutamine residues (Q) on human growth hormone (hGH), only 2 sites, Q141 and Q40, can be transglutaminated by mTGase with Q141 as the preferred site. To obtain a higher yield of a homogeneous hGH product with PEGylated Q141 for pharmaceutical application, we aimed to generate a mTGase mutant library and develop a high-throughput screening (HTS) method for an mTGase with a superior specific activity on Q141 of hGH.
Synthetic poly-glutamine was reported to be used as the mTGase substrate in the screening for TGase mutants of high activity. 14,15 However, screening for mTGase that prefers Q141 of hGH requires the use of hGH as the substrate, in which free amine groups (ε-NH2 group of lysine) can be undesirably catalyzed and thus complicate selectivity measurements. In addition, cellular proteins from Escherichia coli, the expression host of recombinant mTGase, can also be catalyzed as the substrate of mTGase; therefore, high-throughput purification of mTGase becomes essential.
Another challenge for developing the screening assay is the extremely low level of active mTGase that can be directly expressed in E. coli due to the toxicity of TGase to the expression host. Although several methods can bypass this problem by relying on expression of an inactive pro-mTGase (mTGase with pro-domain at its N-terminus) followed by in vitro activation via proteolytic digestion, 14-16 the cost of proteases and automation difficulties impede the adaptation of such strategies.
In this study, we developed a convenient screening method for highly specific mTGases from an mTGase mutation library and identified 5 mTGase mutants with greatly improved specificity toward Q141 of hGH. This method overcomes the bottlenecks of low expression level of mTGase in E. coli and the nonspecific catalytic activity of mTGase using target protein, hGH, as the substrate. It is highly sensitive, easily automated, and applicable to screening for mTGases with selected specificity or activity toward different therapeutic proteins.
Materials and Methods
Microbial strain
Streptomyces ladakanum was obtained from ATCC (American Type of Culture Collection), #27441.
Plasmid construction
pACYCDuet-RL27-3C expressing 3C-protease
DNA sequence encoding 3C protease, Human Rhino Virus strain 14 (HRV14) 3C protease, was cloned into pACYC-Duet1 expression vector (Novagen, Madison, WI) between NdeI and AvaI sites.
pET39b-pro-(3C)-mTGase-(GS)-6xHis
A fragment (3C) encoding 3C-protease (derived from SRV14) digestion site, 17 LEVLFQGP, which cleaves between Q and G, was cloned between pro and mature mTGase domains, resulting in extra GP residues at the N-terminus of the mature mTGase after cleavage. Also, a 6xHis tag was cloned to the C-terminus of mTGase ( Fig. 1 ). For E. coli expression, the DNA sequence encoding pro-(3C)-mTGase-(GS)-6xHis was cloned between NdeI and BamHI sites of pET39b (Novagen) expression vector.

Amino acid sequence of pro-(3C)-mTGase-6xHis based on wild-type mTGase from Streptomyces ladakanum; shaded: pro-domain; underlined: 3C-protease digestion site; framed: mutation site 62 and site 75 for generating HSmTGase (Y62H_Y75F) and the mutation library; dashed underlined: GS-linker; double-underlined: 6xHis tag.
mTGase mutation library generation
HSmTGase, the Y62H_Y75F Gly-Pro-mTGase mutant from S. ladakanum with improved specificity over the wild-type mTGase (data not shown), was used as the template for a small mutant library construction. Mutations were generated at sites 62 and 75 on the template pro-3C-HSmTGase-6xHis. This library contained 29 mTGase mutants, including 17 replacements at site 62 (H62A, C, D, E, F, G, H, I, K, L, P, Q, R, S, T, V, W) and 12 at site 75 (F75A, C, D, I, K, L, M, N, P, R, S, T), respectively. The wild-type mTGase from S. ladakanum and its mutant, HSmTGase, of which the specificities were well characterized, were used as the controls. The mTGase constructs were cotransformed with the plasmid pACYCDuet-RL27-3C separately into E. coli BL21(DE3) for expression.
96-well expression of in vivo activated mTGase by coexpressing pro-3C-HSmTGase-6xHis variant with 3C-protease
For the expression of active mTGases, cells from −80°C frozen stock or fresh colonies were inoculated into each well of 96-deep-well plates containing 0.2 mL TB medium (Terrific Broth supplemented with required antibiotics, kanamycin at 30 µg/mL, for selection), and then the cells were grown to an optical density of 0.8 at 37°C with shaking at 500 rpm in HiGro (Gene Company Limited) to be used as the seed culture. Then, 50 µL of the seed culture was transferred to a new 96-deep-well plate containing 0.5 mL medium in each well and continuously cultivated under the same condition. The expression of mTGase was induced with 0.02 mM IPTG at OD600 of 2 under 20°C overnight. Cell pellet was collected by centrifugation at 4000 g for 15 min for mTGase purification.
To examine the expression pattern of mTGase before and after the in vivo activation, we performed Western immunoblotting. Anti-mTGase polyclonal antibody generated/purified in house was used as the primary antibody and horseradish peroxidase (HRP)–conjugated goat-antirabbit antibody as the secondary antibody.
Affinity purification of mTGase-6xHis in 96-well filter plate by immobilized metal chelating affinity chromatography
Lysis of the cell
Cell pellet from 0.5 mL culture was resuspended in 200 µL of 1× CelLytic Express (C1990, Sigma, St. Louis, MO) buffer and incubated at 37°C for 30 min. Concentrated imidazole solution was added to each well to a final concentration of 20 mM for the following binding step.
Immobilized metal chelating affinity chromatography (IMAC) purification
For the purification of 6xHis tagged mTGases, 10 µL IMAC resin (Bio-Rad, Hercules, CA) preequilibrated with the binding buffer (20 mM imidazole and 300 mM NaCl in 100 mM phosphate buffer, pH 8.0) was loaded to each well of 96-well filter plates, mixed up with 200 µL cell lysate, and incubated at room temperature for 30 min with shaking at 400 rpm. Then the plates were centrifuged at 1000 g for 5 min and washed twice with 200 µL binding buffer to remove the unbound protein. Purified 6xHis-tagged mTGases were incubated with 25 µL elution buffer (250 mM imidazole and 300 mM NaCl in 100 mM phosphate buffer, pH 8.0) at room temperature for 5 min and then eluted by centrifugation into new 96-well plates.
For optimization of the purification of mTGase in 96-well plates, a different amount (bed volume) of IMAC resin of 50, 40, 30, 20, and 10 µL was used, respectively, to bind with 200 µL cell lysate (prepared from 0.5 mL culture), and then mTGase-6xHis was eluted with 25 µL elution buffer for 3 times.
mTGase activity measurement
One unit of mTGase can catalyze the formation of 1.0 µmol of hydroxamate per minute from Na-CBZ-glutaminylglycine and hydroxylamine at pH 6.0, 37°C. L-Glutamic acid g-monohydroxamate is used as the standard. 18
Preparation of hGH substrate for transglutamination
BirA tag (GLNDIFEANKIEWHE), with biotin ligated to the lysine residue during expression, was cloned to the N-terminal of hGH mutants hGHQ40N and hGHQ141N, respectively, which were expressed in E. coli by coexpressing BirA-hGH mutants with pBIRACm encoding biotin ligase. Biotin was added to the culture medium at the time of induction to a final concentration of 50 µM. Biotin-hGHQ40N and biotin-hGHQ141N were affinity purified with avidin-coupled agarose (Sigma).
Scintillation proximity assay
Tritium-labeled (2,3-3H)-putrescine dihydrochloride, (NH2(CH2)4NH2) at 1.0 mCi/mL, 2.8 µg/mL, in 0.01 N HCl (MT1510; Moravek Biochemicals, Brea, CA), was used as an amine donor.
Streptavidin–scintillation proximity assay (SPA) beads (GE Healthcare Life Sciences, Uppsala, Sweden) were used at 0.5 mg/50 µL/well in 100 mM phosphate buffer, pH 6.0.
SPA reaction
Briefly, in each well of 96-well plates, 1 µg of hGH was premixed with 0.1 µCi of 3H-putrescine in 40 µL of phosphate buffer (100 mM, pH 7.4), and then 10 µL of the purified mTGase, which contained 250 mM imidazole from the purification, was added to start the reaction (each 50-µL reaction solution contains 33 µM [0.1 µCi] 3H-putrescine, 0.82 µM hGH, and 60 mU mTGase). After incubation at room temperature for 16 h, 50-µL suspension of SPA beads was added to each reaction and incubated at room temperature for 30 min before measurement. Count per minute (cpm) from each well was measured with the Micro-Beta plate reader (PerkinElmer, Waltham, MA).
The specificity of mTGase mutant measured by SPA was expressed as cpmhGHQ40N/cpmhGHQ141N.
Confirmation assay by capillary electrophoresis
Transglutamination reaction was performed using 60 µM of the wild-type hGH as the substrate and 90 mM of 1,3-diamino-propanol as the amine donor in 10 mM Tris-HCl (pH 8.0). The reaction was started by adding purified mTGase to a final concentration of 10 µg/mL (0.2 µM) and incubated at room temperature. Samples were collected at time intervals (0-24 h) and immediately frozen with liquid nitrogen until the analysis by capillary electrophoresis (CE; P/ACE MDQ, Beckman Coulter, Fullerton, CA). From the CE profiles, the retention times for wild-type hGH, mono-substituted hGH at Q141 (Q141_derivitive), and mono-substituted hGH at Q40 (Q40_derivitive) were 6.5, 7.9, and 10 min, respectively. The specificity of mTGase was expressed as the ratio of the amount of Q141_derivative to Q40_derivative.
The amount of conjugated or unconjugated hGH was measured by CE. The hGH conversion rate was calculated as the ratio of the amount of hGH reacted to the total amount of hGH added to the reaction.
Results and Discussion
Identification of the highly specific mTGase as the template for mutation library generation
To generate an mTGase mutation library and screen for mutants that favor the conjugation of PEG to Q141 than Q40 of hGH, we desired to use a template that already had high specificity toward Q141. During the process development for mTGase production, mTGase variants with different N-termini were generated from different in vitro activation processes. For example, Met-mTGase was expressed by E. coli, Ala-Pro-mTGase was generated from the enterokinase cleaved pro-mTGase, and Gly-Pro-mTGase was from the 3C-protease (SRV14-derived) cleaved pro-(3C)-mTGase. The specificities of these N-terminal variants were compared with that of the wild-type at the point when 40% of hGH was converted, and the results in Table 1 show that Gly-Pro-mTGase ( Fig. 1 ) had the highest specificity of 8.8 compared to 5.6 of the wild-type. This clearly indicated that the N-terminal sequence of mTGase affected the specificity of this enzyme.
Comparison of the Specificity of mTGase Variants Derived from Streptomyces ladakanum Measured by Capillary Electrophoresis

The specificity was calculated as the amount of hGHQ141_derivative over hGH40_derivative when about 40% of hGH was converted. Met-mTGase was expressed by Escherichia coli. Ala-Pro-mTGase was generated by enterokinase digestion on pro-mTGase. The Gly-Pro-mTGase and its Y62H_Y75F mutant (HSmTGase) were generated by 3C-protease digestion of pro-(3C)-mTGase. Wild-type hGH was used as the substrate, and 1,3-diamino-propanol was used as the amine donor.
Moreover, according to the published data of the crystal structure on mTGase by Kashiwagi et al. 19 and Shimba et al., 20 Y62 and Y75 sites are among the critical residues that can affect the mTGase activity. We generated site-saturated mutations on Y62 and Y75 separately and found that both Y62H and Y75F mutants of mTGase had increased specificities toward the Q141 site of hGH. Further increased specificity was observed when combined Y62H and Y75F within 1 mutant. On top of these observations, we wanted to know if a different combination of the mutants on sites 62 and 75 would give a higher specificity on Q141 of hGH regardless of activity and generated the small library in this study, as described in Materials and Methods.
HSmTGase, the mutant containing the point mutations of Y62H and Y75F of Gly-Pro-mTGase (Gly-Pro-mTGase_Y62H_Y75F), had a superior specificity of 18.8 toward hGHQ141, which was 3 times higher than 5.6 of the wild-type mTGase. This indicated that the amino acids at sites 62 and 75 also affected the specificity of mTGase. Therefore, we generated a mutation library by randomly replacing the amino acid at site 62 or 75 of HSmTGase for screening.
Achievement of a high level of active mTGase expressed in E. coli
A common procedure for producing high levels of mTGases in E. coli and avoiding mTGase-induced cytotoxicity to the expression host involves first expressing and purifying the inactive pro-mTGase and then activating them by in vitro protease cleavage to remove the pro-domain. 14 However, it is very challenging to produce all of the mutants made with this approach to support a high-throughput screen of mTGases because of the low efficiency. To yield sufficient mTGases for the transglutamination assay, we developed an in vivo activation method to express high levels of active mTGases directly in E. coli. This was achieved by coexpressing pro-(3C)-mTGase-6xHis with 3C-protease in E. coli, in which the N-terminal pro-domain was processed by 3C-protease within the cell. E. coli was grown in TB medium and induced with an extremely low level of IPTG at 0.02 mM at a low temperature of 25°C overnight. High expression levels of active mTGases around 0.2 U/mL/OD600 were achieved, which was >10-fold higher than that directly expressed from E. coli (data not shown).
Next we wanted to evaluate the efficiency of in vivo processing of pro-(3C)-mTGase by Western immunoblotting of the cell lysate from the coexpression culture. Cell lysate from expressing pro-(3C)-mTGase-6xHis alone was used as the control. Results in Figure 2 show that pro-(3C)-mTGase-6xHis produced by cotransformed E. coli was completely processed to the smaller protein, which lacked the pro-domain (lane 2), whereas pro-(3C)-mTGase-6xHis produced in the control remained unprocessed (lane 1; Fig. 2 ). This indicated that coexpressed 3C-protease efficiently cleaved the nascently produced pro-(3C)-mTGase-6xHis to generate the active mTGase. Although active mTGase can be expressed alone in E. coli, the cytotoxicity associated with mTGase activity forbids high expression to meet the transglutamination assay sensitivity. In this study, we report for the first time that a high level of active mTGase can be expressed directly from E. coli, which makes HTS of mTGase mutants possible.
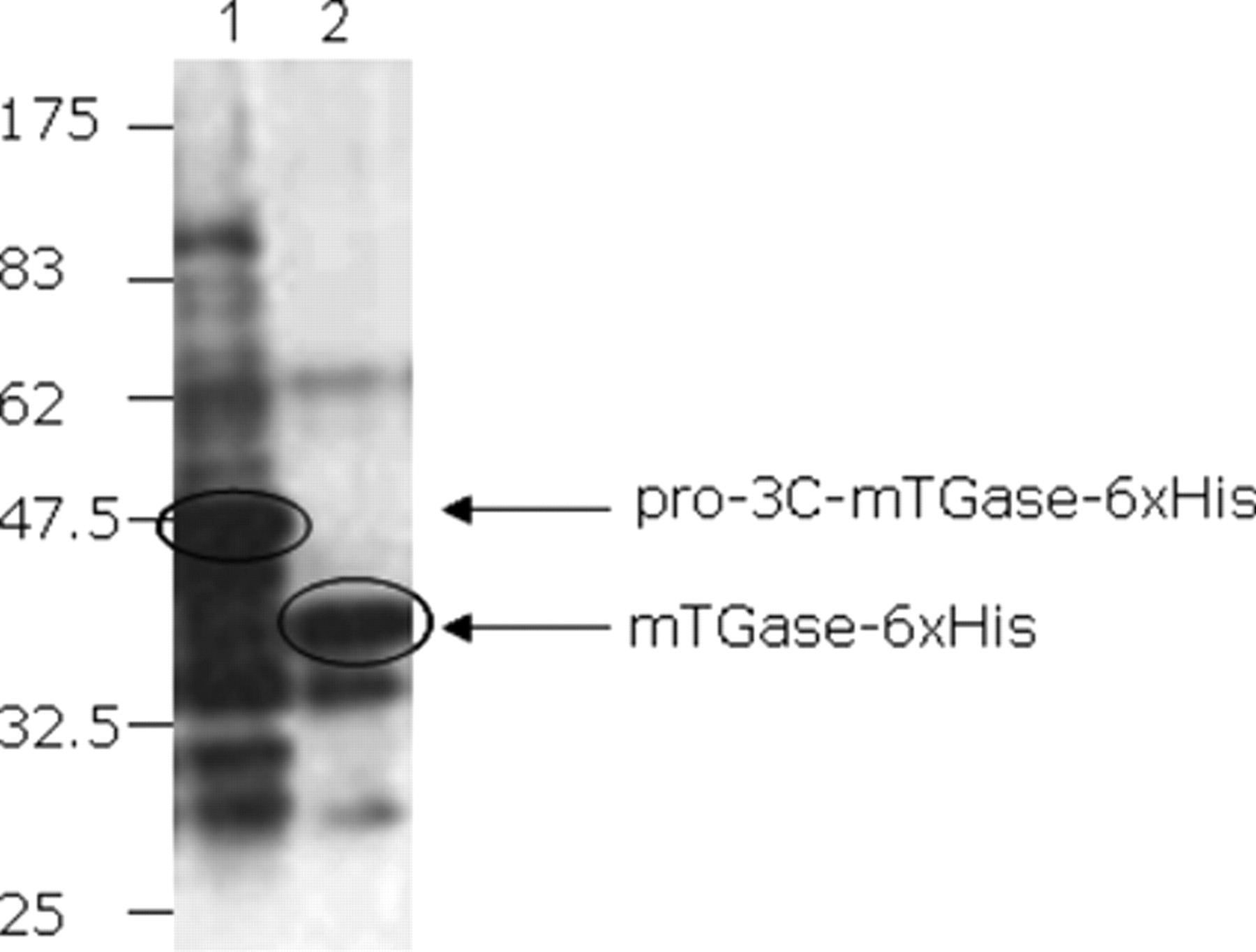
Evaluation of the in vivo activation of mTGase by Western immunoblotting using anti-mTGase polyclonal antibody. Lane 1: cell lysate from Escherichia coli expressing only pro-3C-mTGase-6xHis; lane 2: cell lysate from E. coli coexpressing 3C-protease and pro-3C-mTGase-6xHis. No pro-3C-mTGase-6xHis was observed in lane 2, indicating the efficient processing of active mTGase-6xHis by 3C-protease.
The Western immunoblotting result also demonstrated that the expression of pro-(3C)-mTGase-6xHis alone resulted in strong degradation and dimerization of the target protein (lane 1, Fig. 2 ), which was highlighted by the anti-mTGase polyclonal antibody, whereas coexpression with 3C-protease can greatly reduce these undesired products (lane 2, Fig. 2 ).
Establishment of high-throughput affinity purification of mTGase-6xHis in a 96-well plate format
Because more than 50 mM imidazole in the transglutamination reaction will interfere with mTGase activity (data not shown), maximally 10 µL of mTGase-6xHis eluted with 250 mM imidazole is allowed in each 50-µL transglutamination reaction. Therefore, for optimization of the purification procedure, we aimed at obtaining the highest recovery of mTGase-6xHis from the first elution fraction using the 25-µL elution buffer. For this purpose, different amounts (bed volume) of IMAC resin of 50, 40, 30, 20, and 10 µL were used, respectively, to bind with 200 µL cell lysate (prepared from 0.5 mL culture), and then mTGase-6xHis was eluted with the 25-µL elution buffer 3 times. The recovery of mTGase-6xHis from each elution fraction was evaluated by the recovery of mTGase activity ( Table 2 ) and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; Fig. 3 ), respectively. As the ratios of Volumeelution-buffer/ Volumeresin were increased from 0.5 (using 50 µL resin) to 2.5 (using 10 µL resin), mTGase recovered in the first fraction also increased, reaching the maximal recovery rate of 66.4% at 10 µL resin ( Table 2 ). This was consistent with the results evaluated by SDS-PAGE shown in Figure 3 that the highest recovery of mTGase-6xHis in the first fraction was obtained by using 10 µL resin (10 µL resin/fraction 1).
Recovery of mTGase Activity in a 96-Well Affinity Purification Format from 0.5 mL Culture Using Different Amounts of IMAC Resin of 50, 40, 30, 20, and 10 µL
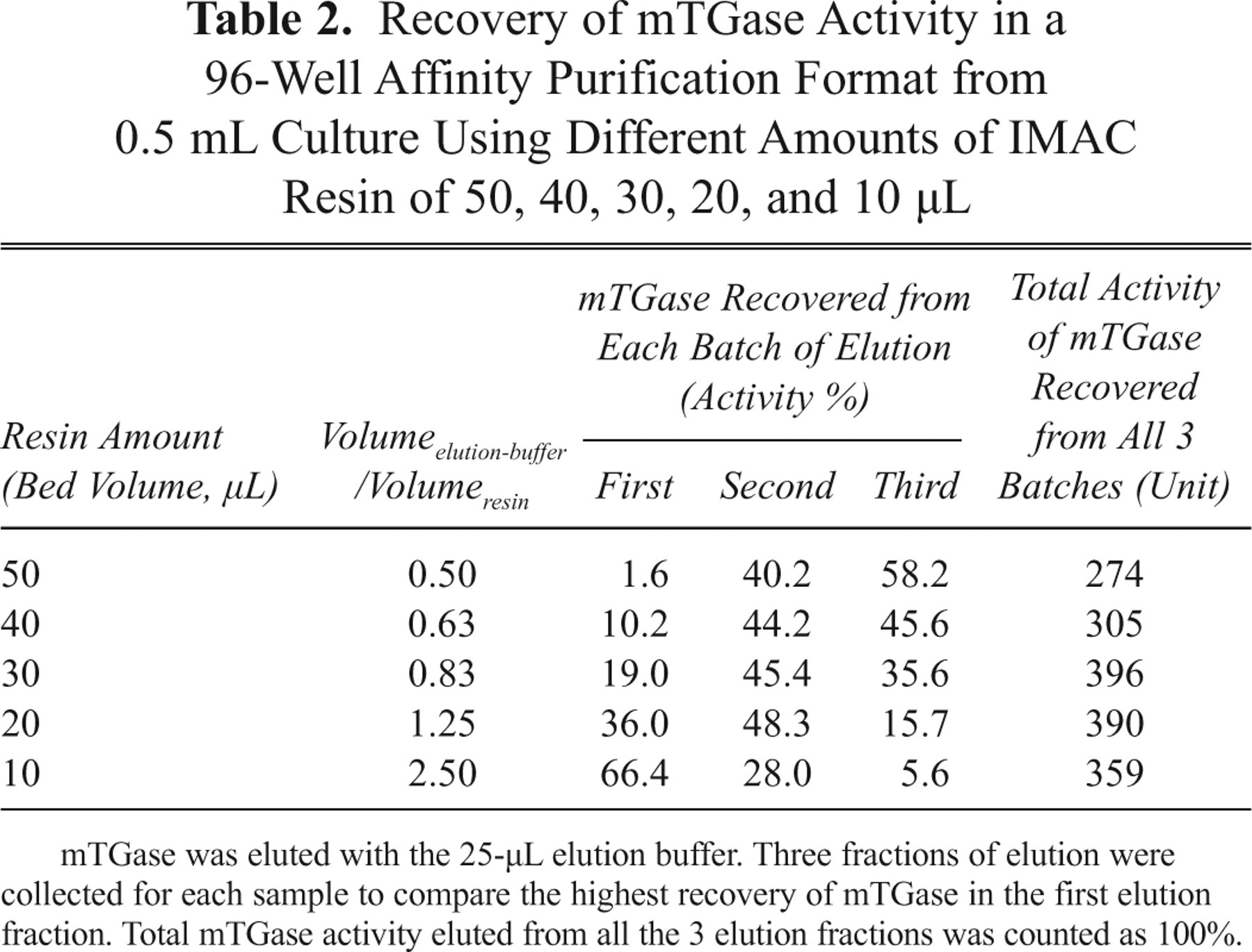
mTGase was eluted with the 25-µL elution buffer. Three fractions of elution were collected for each sample to compare the highest recovery of mTGase in the first elution fraction. Total mTGase activity eluted from all the 3 elution fractions was counted as 100%.
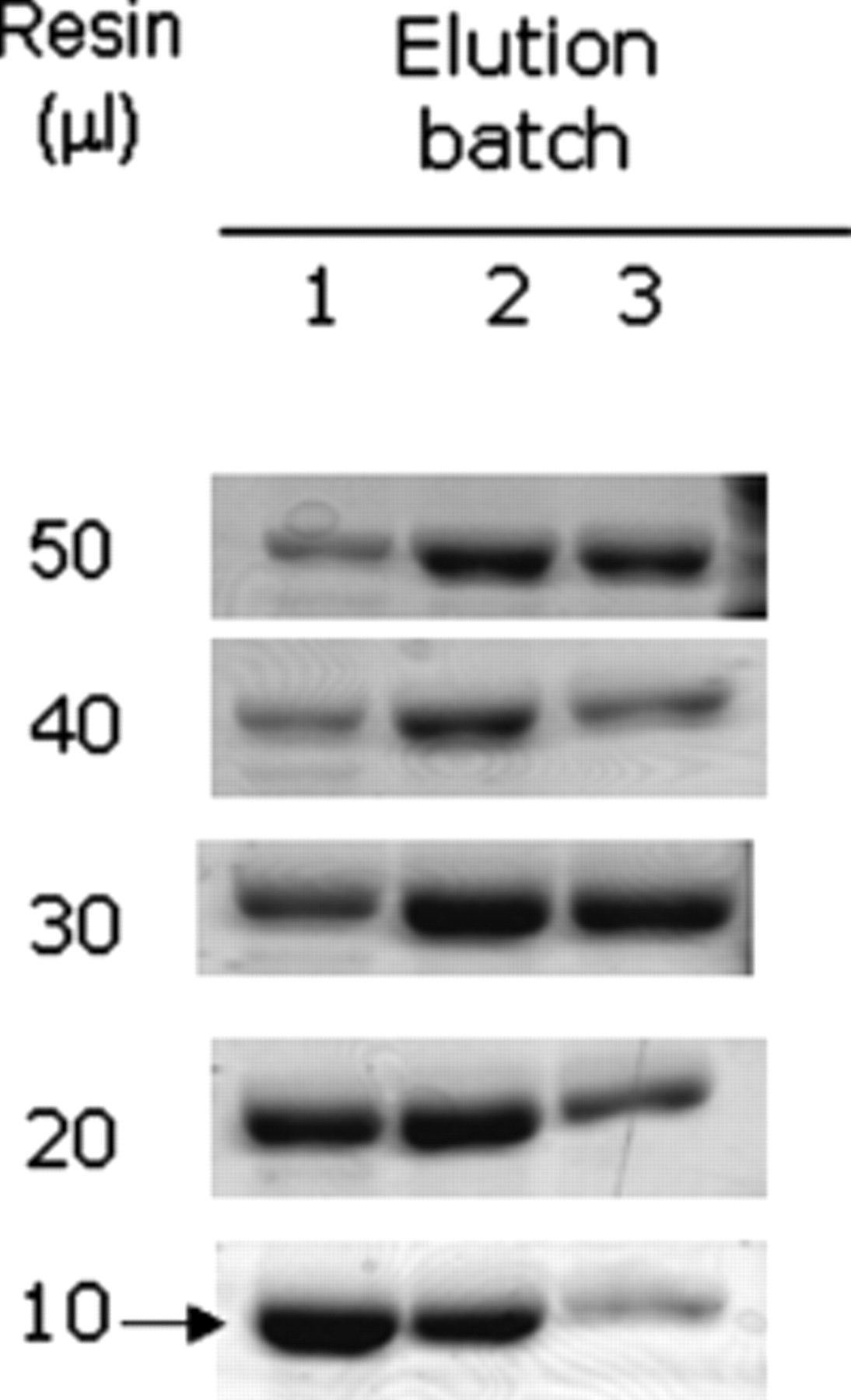
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) showing 96-well purification of HSmTGase-6xHis from 0.5 mL culture using different amounts of immobilized metal chelating affinity chromatography (IMAC) resin from 50, 40, 30, 20, and 10 µL. Each sample was eluted with the 25-µL elution buffer for 3 batches. Only the first elution fraction was of interest for the transglutamination assay.
To use the active mTGases directly in the screening assay, we needed to normalize mTGase at the protein level during the purification step. The total recovery of mTGase in Table 2 showed that 390 units were recovered by 20 µL of resin, whereas only 359 units were captured by 10 µL of resin, implying that the amount of mTGase from the 0.5-mL culture that contained >390 units could oversaturate 10 µL of resin ( Table 2 ). Therefore, by saturating the binding capacity of 10 µL of resin with an exceeding amount of mTGase from the 0.5-mL culture, we could normalize mTGase at protein levels. The normalization of mTGase using 10 µL of resin was further evaluated by the purification of the 29 mTGase mutants and the 2 controls, WT (wild-type) and HSmTGase. The amount of mTGase eluted in the first fraction was analyzed by SDS-PAGE ( Fig. 4 ). It was shown that similar amounts of mTGase-6xHis protein were eluted for most of the variants, indicating that the mTGase-6xHis protein was successfully normalized by limiting the amount of IMAC resin. Interestingly, 3 mutants—75D, 62D, and 62P—had lower recovery than the others, perhaps because of the low level or insoluble expression of mTGase. However, this had no effect on selecting other high-specific mutants.
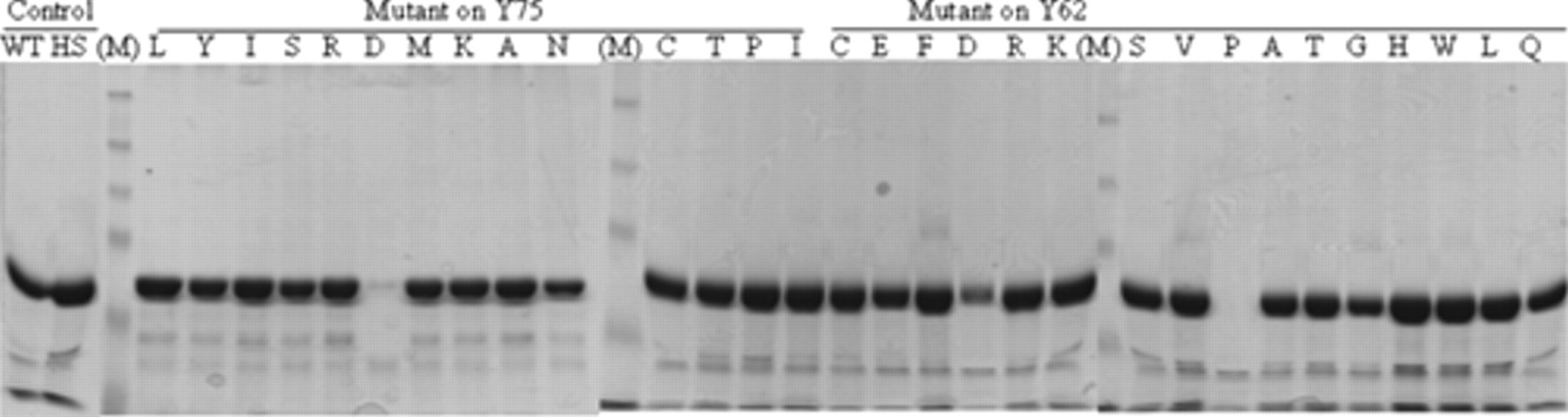
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the normalization of 31 mTGase variants, including 29 mutants and 2 controls at the protein level by limiting the amount of immobilized metal chelating affinity chromatography (IMAC) resin (10 µL) in 96-well purification. WT, wild-type mTGase-6xHis; HS, HSmTGase-6xHis; (M), prestained molecular weight marker (New England Biolabs, Ipswich, MA).
Identification of 5 mutants with improved specificity by the SPA-based screening assay
The mTGase variants including the mutants and controls were tested in 3 independent experiments starting from expression, purification, to the SPA-based transglutamination assay. The mutants that showed improved specificity while retaining >50% activity of HSmTGase were chosen as the candidates for subsequent confirmation assay. Five mutants—62A, 62C, 62L, 62S, and 75N—were selected to have better specificity than that of HSmTGase ( Fig. 5 ).
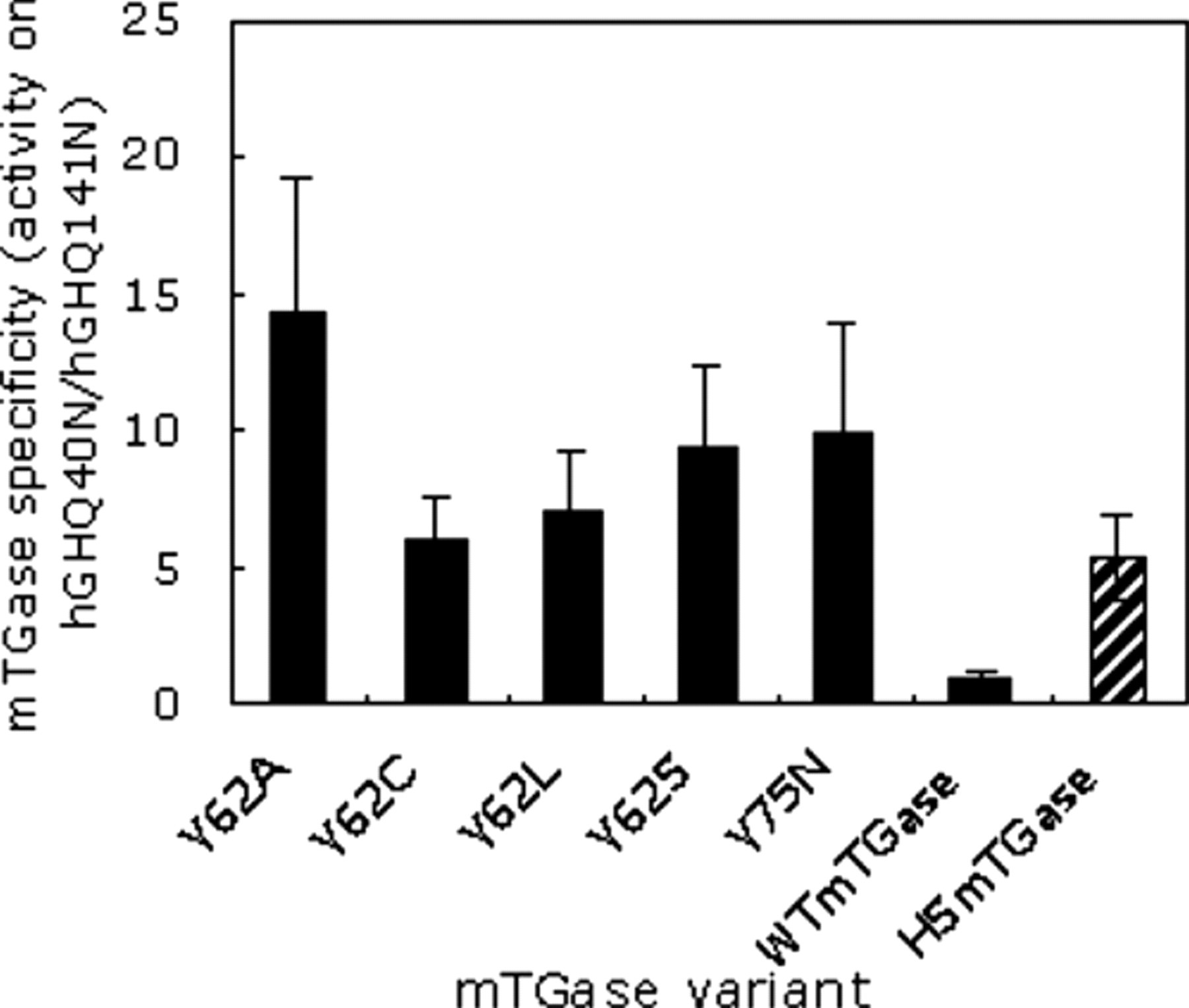
Comparison of the specificity of the 5 mutants obtained from the high-throughput screening with that of WTmTGase or HSmTGase. The data were the averages from 3 different runs, which started from the inoculation for expression and then went through purification and screening. For each run that we had triplicates, the data for these triplicates had a p-value < 0.05.
The SPA-based high-throughput assay for mTGase activity using synthetic glutamine-containing oligo peptide has been reported. 21 Instead of using the reported 4 µCi of (2,3-3H)-putrescine/reaction, we used a much lower amount at 0.1 µCi of (2,3-3H)-putrescine/reaction in this study, which proved to be more economical and environmentally friendly.
Confirmation of the high specificity by CE analysis
To confirm the high specificity of the 5 mutants obtained from the screening, we carried out transglutamination reaction using wild-type hGH as the substrate and quantified the hGH derivatives modified on Q141 (Q141_derivative) and Q40 (Q40_derivative), respectively, by CE. The generation of Q141_ or Q40_derivative catalyzed by 62A, 62L, WTmTGase, and HSmTGase over time was compared ( Fig. 6A , B ). At 16 h of reaction, almost no Q40_derivative, which was undesired, was generated by either 62A or 62L, whereas WTmTGase and HSmTGase catalyzed 19% and 12% of the total hGH to become Q40_derivative, respectively. In addition, with prolonged reaction time, modification on both Q141 and Q40 sites would be observed for the 2 control mTGases. This indicated that the 2 mutants, 62A and 62L, had exclusively high specificity toward Q141 of hGH. Data for the other 3 mutants—62C, 62S, and 75N—were very much similar to those for 62A and 62L (data not shown). Most important, this result confirmed the high specificity of the mutants obtained from the screening assay, indicating the effectiveness of the screening method we described in this study.
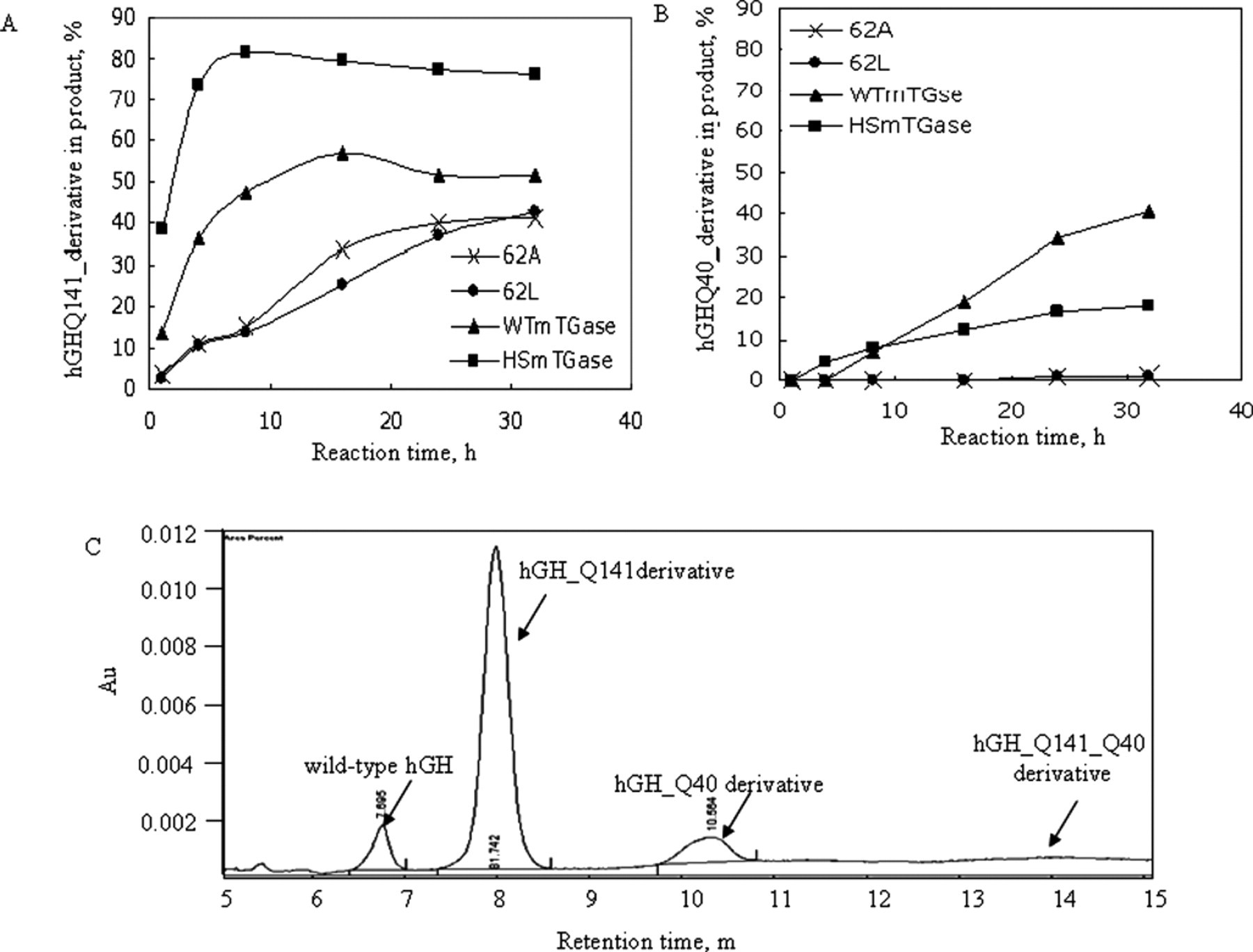
Confirmation of the improved specificity of the mutants, 62A and 62L, by capillary electrophoresis (CE). (
The results in Figure 6A also indicated that the reaction rate for both WTmTGase and HSmTGase was much faster than that of the mutants. The slow reaction rate for the mutants allowed the reaction exclusively on Q141 at least within 16 h of the reaction, thus preventing the generation of the unwanted product Q40_derivative. This could avoid the extra downstream separation of Q40_ and Q141_derivatives in the final product, which would be difficult because of the high similarity between the 2 derivatives. Therefore, we successfully screened mTGase mutants that had both high specificity and desired reaction rate that was applicable for industrial applications. Our screening system also allows stopping the reaction at a desired reaction time to compare specificity of the mutants. Because different mutants have different reaction rates, the ideal way to screen for highly specific mTGases is to compare the kinetics of each mutant. Yet so far, an HTS method has not been available to meet such criteria.
The profile of the remaining hGH and the generated derivatives at 16 h of reaction by HSmTGase is shown in Figure 6C . The modification on both Q141 and Q40 sites was not observed until after 16 h or even longer, which may have been caused by the mTGase mutants possessing high Q141 specificity that had restricted accessibility for Q40 of hGH.
Conclusions
In this study, we obtained 5 mTGase mutants that were highly specific toward Q141 of human growth hormone via a novel HTS method, which is reported for the first time to screen for highly specific mTGase mutants toward the desired site of a therapeutic protein. The method was developed by (1) achieving a high expression level of active mTGase directly from E. coli, (2) establishing 1-step purification of mTGase, and (3) using therapeutic protein (hGH in this case) as the transglutamination substrate. More important, we achieved high sensitivity by using a much lower level (0.1 µCi) of tritium-labeled amine donor, 3H-putrescine. The method is highly sensitive, inexpensive, and easily automated, and it can be applied to screening customized mTGase with desired specificity or activity targeting different therapeutic proteins.