
Abstract
COVID-19 (Coronavirus Disease 2019) is a highly contagious infection and associated with high mortality rates, primarily in elderly; patients with heart failure; high blood pressure; diabetes mellitus; and those who are smokers. These conditions are associated to increase in the level of the pulmonary epithelium expression of angiotensin-converting enzyme 2 (ACE-2), which is a recognized receptor of the S protein of the causative agent SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2). Severe cases are manifested by parenchymal lung involvement with a significant inflammatory response and the development of microvascular thrombosis. Several factors have been involved in developing this prothrombotic state, including the inflammatory reaction itself with the participation of proinflammatory cytokines, endothelial dysfunction/endotheliitis, the presence of antiphospholipid antibodies, and possibly the tissue factor (TF) overexpression. ARS-Cov-19 ACE-2 down-regulation has been associated with an increase in angiotensin 2 (AT2). The action of proinflammatory cytokines, the increase in AT2 and the presence of antiphospholipid antibodies are known factors for TF activation and overexpression. It is very likely that the overexpression of TF in COVID-19 may be related to the pathogenesis of the disease, hence the importance of knowing the aspects related to this protein and the therapeutic strategies that can be derived. Different therapeutic strategies are being built to curb the expression of TF as a therapeutic target for various prothrombotic events; therefore, analyzing this treatment strategy for COVID-19-associated coagulopathy is rational. Medications such as celecoxib, cyclosporine or colchicine can impact on COVID-19, in addition to its anti-inflammatory effect, through inhibition of TF.
Introduction
In December 2019, there was a new highly contagious infectious disease outbreak in Wuhan, China. Some infected patients developed severe acute respiratory syndrome (SARS) and a systemic inflammatory response syndrome (SIRS) associated with a high mortality rate. This disease then rapidly spread throughout the world and a pandemic was declared in March 2020. 1 The causative agent of the novel disease known as COVID-19 (Coronavirus Disease 2019) was isolated and identified as a new coronavirus, known as SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2). 2 Studies report that S protein from the virion surface binds to the angiotensin-converting enzyme 2 (ACE-2) expressed in respiratory epithelial cells, thus triggering mechanisms that cause the virus enter the cell where it manages to generate its replicants. 3 At least 2 types of biological response have been identified and are related to the pathogenesis of the disease: the induction of an inflammatory response and the generation of a procoagulant state.
The immune/inflammatory response can be mild and rapidly self-limiting. Where the immune/inflammatory response is mild and rapidly self-limiting, patients will present as asymptomatic or with only mild symptoms. Patients with less severe disease symptoms achieve, as far as is known, both cellular and humoral immunity for protection against future virus exposures. 4 This immune response may be poorly regulated in other infected individuals, progressing to SARS and SIRS, both of which are associated with high mortality rates. 5 Moreover, many of these patients develop thrombotic coagulopathy, 6 which is clinically manifested as cerebrovascular accidents, 7 acroischemia, 8 or pulmonary thromboembolism. 9 The presence of thrombi in pulmonary microcirculation is the most common result when performing an autopsy (27%). 10 The most common abnormalities in hemostatic screening tests are prolonged prothrombin times (PT) and partial thromboplastin times (PTT), elevated D-dimer serum levels, and reduced fibrinogen and platelet count levels. 11 Thrombin, tissue plasminogen activator (tPA), tissue factor pathway inhibitor (TFPI) and vascular endothelial growth factor (VEGF) significantly more increased in the critical versus noncritical patients. 12
COVID-19 severity has been associated with advanced age and comorbidities such as chronic obstructive pulmonary disease (COPD), heart failure, hypertension (HTN), diabetes mellitus, and smoking. 13 These comorbidities have been associated with pulmonary ACE-2 overexpression; moreover, this group of viruses causes ACE-2 down-regulation, resulting in increased angiotensin 2 (AT2). 14 This acute increase in levels of AT2 may have direct implications for the immune, vascular endothelial and coagulation responses. 15,16
Accumulation of AT-II secondary to ACE2 downregulation may promote clot formation via interactions with endothelial cells and platelets 17 ; TF may be involved in this process. 18 These findings may also be associated with additional prothrombotic events that have been identified in patients diagnosed with COVID-19, including increases in serum IL-6 which has characterized activities with respect to coagulation factors, 19 and platelets. 20 IL-6 can activate directly TF. 21 T lymphocytes as well as the cytokines IL-6, and TNF-α may have direct roles in promoting microvascular damage associated with AT-II. 22 –24
Neutrophil extracellular traps (NETs) are extracellular webs of chromatin, microbicidal proteins, and oxidant enzymes that are released by neutrophils to contain infections, and have potential to propagate inflammation and microvascular thrombosis. Sera from individuals with COVID-19 triggered NET release from control neutrophils in vitro. 25 Interestingly IL-1β mediates arterial thrombus formation via NET-associated TF. 26
It’s important to put attention on generation of proinflammatory cytokines and vascular inflammation are mediated through a number of pattern recognition receptors, known as toll-like receptors (TLRs) and nod-like receptors (NLRs). 27
Activation of TLRs through oxidized phospholipids or damage associated molecular pattern (DAMP) can activate the production of TNFα, IL6, and other cytokines that are responsible for cytokine storm in COVID-19. 28 Interestingly, one in silico interaction study hypothesized that TLR4 may be involved in recognizing molecular pattern from spike protein of coronavirus. Further studies will be required to understand the roles of TLRs in COVID-19. 29
Both an acute inflammatory response 30 and increased AT2 31 are known causes for TF overexpression and the activation of the blood clotting cascade, an event that has been associated with COVID-19 pathogenesis. TF upregulation may possibly participate in the pathogenesis of the disease 32 -34 as part of a chain of events where endothelial dysfunction/endotheliitis are involved, 35 Willebrand factor and soluble thrombomodulin overexpression 36 and ADAMTS13 deficiency (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). 37,38 Unmasking these pathways in COVID-19 would have a key role in designing therapeutic strategies. On the other hand, the increase in AT2 can cause vasoconstriction with increased blood pressure, which can lead to hemorrhagic stroke. 39 In some patients with COVID-19, significant deposits of terminal complement components C5b-9 (membrane attack complex), C4d, and mannose binding lectin (MBL)-associated serine protease 2 (MASP-2) have been found in the microvasculature of different organs. 40
This accumulated observation of the activation of the complement system in COVID-19, it is quite likely to contribute in developing thrombotic processes, 41 –43 as previously assessed from the physiological interplay of complement and blood clotting cascades. 44 The role of TF activation induced by activated complement factors such as C5a through the effect of protein disulfide isomerase (PDI) 45 should be examined in COVID-19, which could be the basis for developing therapeutic proposals.
Protein C, a natural anti-coagulant is reduced in patient with COVID-19. 46 Activated protein C is a prominent anti-coagulant, anti-inflammatory molecule that regulate endothelial dysfunction and TF expression in monocyte could be useful therapeutically. 47
Figure 1 summary as immunological dysfunction, ACE2 downregulation, and AT2 and TF upregulation would be involved in the thrombosis pathogenesis in COVID-19.
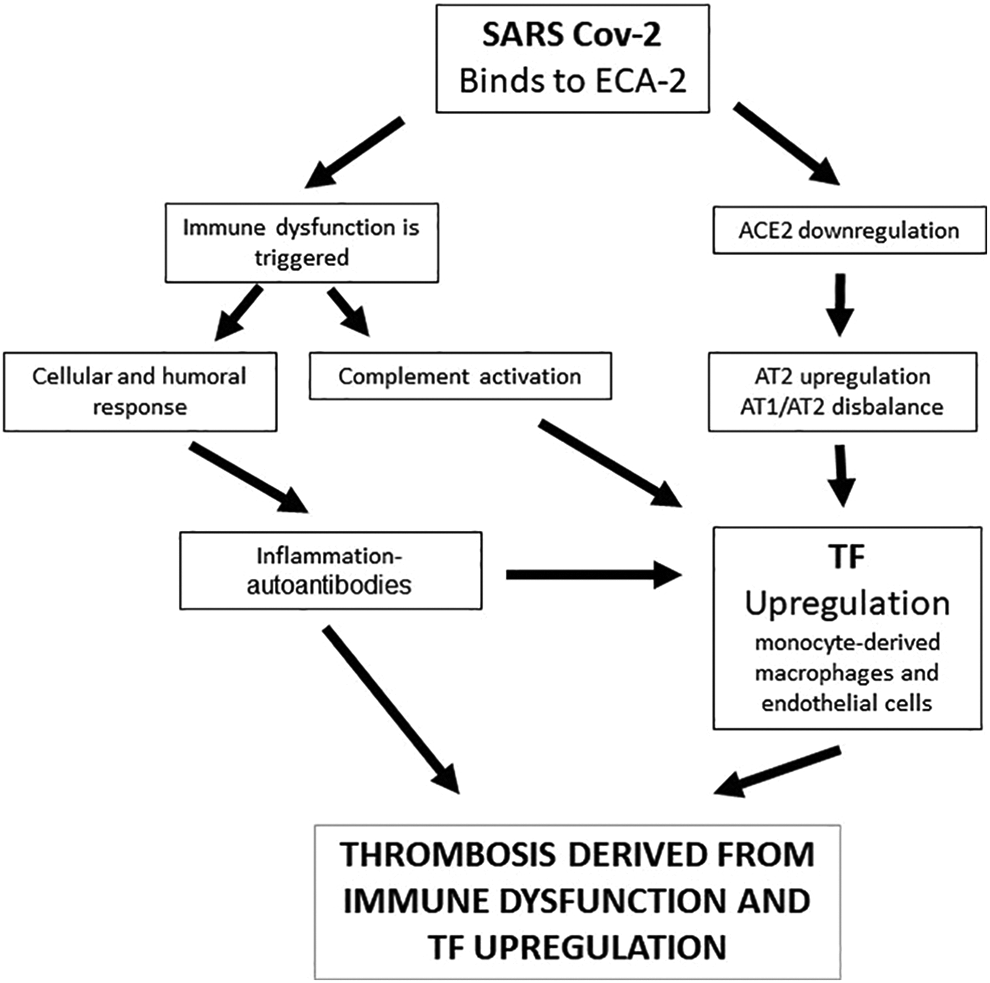
Summary of the possible mechanisms associated with thrombosis in COVID-19 where immunological dysfunction, angiotensin-converting enzyme 2 (ACE2) downregulation, and angiotensin 2 (AT2) and tissue factor (TF) upregulation would be involved.
Tissue Factor (TF)
TF was discovered in 1904 as a protein that converted prothrombin to thrombin at the plasma level. 48 Being a transmembrane protein with a high affinity for Factor VII and Factor VIIa, it is known as thromboplastin, coagulation factor III or CD 142; moreover, it acts as an initiator of the blood clotting cascade by activating Factors IX 49 and X. 50 It first appeared in animals 450 million years ago in the evolutionary path of agnate fish. 51 The gene is found on chromosome 1 and has 6 exons. 52
It occurs dimerically, with 2 extracellular domains belonging to the immunoglobulin superfamily, the intracellular part is bound to intracellular Jak-STAT signaling structures, being classified within the group of type II cytokine receptors. 53,54 It emits intracellular responses that lead to compartment-specific changes in calcium, 55 with biological cell migration and proliferation effects, 56 as well as angiogenic effects at embryonic stages. 57
TF contains an allosteric disulfide bond that stabilizes the carboxyl-terminal domain involved in ligand interactions with coagulation factors VIIa and X. PDI is involved in modifying allosteric disulfide by reduction to achieve the activation thereof (change from an encrypted to a decrypted conformation of TF). 58,59
TF is mainly expressed in endothelial cells, 60 platelets, 61 polymorphonuclear cells, 62 T lymphocytes, 63 and fibroblasts. 64 It is located mainly in lipid rafts 65 and closely linked to β1 integrins. 66
The main physiologic trigger for TF activation (transition from an inactive encrypted to a decrypted conformation) is tissue injury that results in hemostasis. This activation primarily occurs in the endothelium, platelets, and perivascular cells. 67 Under pathological conditions, it is released into the blood as vesicles or microparticles. The release of microparticles has been observed in conditions where there are thrombotic events, from fractured atheromatous plaque (at the level of Foam cells), monocytes in septic states, or neoplastic cells. 68 –70
Different TF gene agonists in endothelial cells have been studied, including tumor necrosis factor alpha (TNFα), 71 interleukin-1β (IL-1β), 72 CD40 L, 73 vascular endothelial growth factor (VEGF), 74 serotonin, 75 histamine, 76 and C-reactive protein. 77 Stimulated P-selectin increases monocyte TF expression. 78 The induction of intracellular signaling is essential in the TF activation process. 79 –81
Anti-cardiolipin antibodies and anti-β2-glycoprotein 1 increase TF expression in monocytes in antiphospholipid syndrome. 82,83 Vitamin D can modulate TF expression in these patients. 84
TF Expression Regulation
The TF activation process is regulated at both transcriptional and post-transcriptional levels. 85 The positions of various membrane phospholipids that generate different polarities, 86 the PDI effect on disulfide bonds, 87 and the TFPI (tissue factor pathway inhibitor) effect of the inhibitory protein 88 are all involved. Hemostasis requires a balance between TF and TFPI.
These pathways converge at a common point in the complement system. When activated, factors such as C5a can activate the TF by modifying its sulfhydryl radicals via PDI mediation. 89
Anti-inflammatory cytokines such as IL-4, IL-10 and IL-13, 90 –93 as well as prostaglandin (PG) E1 and PGI2, can suppress TF in monocytes and macrophages. 94
TF in Viral Infections
The coagulation system can be activated via TF during viral infections such as the Herpes simplex virus, 95 the human immunodeficiency virus (HIV), 96 the Coxsackievirus B3, 97 dengue, 98 or Ebola. 99 This response most likely evolved as a host defense system to prevent the virus from spreading. However, acute viremia can lead to a type of consumptive coagulopathy, thus contributing to multiorgan failure and death. 100 The activation of toll-like receptor 3 (TLR3) in the presence of double-stranded RNA viruses can induce TF expression in cultured endothelial cells and activate the coagulation system in mice. 101 Note that the inhibition of the factor VIIa-tissue factor complex reduced cytokine storm and mortality in a Rhesus macaque model of Ebola hemorrhagic fever. 102
TF in COVID-19
Three recent reviews speculate that induction of TF expression may play a role in COVID-19-related thrombosis. 32 –34 The correlation between TF expression and the severity of COVID-19 has been studied both its expression in monocytes 103 and in extracellular vesicles. 104 Platelets from severe COVID-19 patients were able to induce TF expression ex vivo in monocytes from healthy volunteers. 105 TF can increase its expression during hypoxia and generate a thrombotic tendency; phenomenon that is mediated through extracellular RNA activated Toll-like receptor 3-activated protein 1 signaling. 106
It is too early to conclude at which point in the cascade of biological events triggered in COVID-19 this TF overexpression is found; later studies will surely clarify it for us.
TF-Specific Inhibitors
There are vast therapeutic strategies focused on reducing the blockade of TF expression in prothrombotic conditions and it is the subject of various studies. 107
There are 5 types of TF-specific inhibitors that are currently being examined to be medically administered as antithrombotic agents: monoclonal antibodies against TF, 108 recombinant TFPI, Ixolaris (protein identified in the tick saliva with 2 Kunitz-like domains), 109,110 molecules that interfere with the TF binding site with factor VIIa 111 and anticoagulant nematode protein C2. 112
Other Medications That Can Modulate TF Expression
Various drugs for regular use in different fields of medicine have been studied for their potential inhibitory role in TF expression. Published studies have been of a different nature, including in vitro, in vivo, and ex vivo studies, and usually report the intracellular mechanisms of action involved. Some of the drugs studied are angiotensin-converting enzyme 1 (ACE-1) inhibitors, 113 antioxidants, 114 clopidogrel, 115 salicylates (acetylsalicylic acid, ibuprofen, indomethacin), 116 specific COX-2-specific inhibitors (celecoxib), 71 cyclosporine A, 117 hydroxyurea, 118 vitamin D, 119 pentoxifylline, 120 and Omega-3. 121 Interestingly, glucocorticoids such as dexamethasone increase TF induction in LPS-stimulated monocytes have little effect on endothelial cells. 122
Another source of TF activation inhibition may be through the suppression of the complement pathway, as could eculizumab, a C5-specific inhibitor, on which studies for COVID-19 are even being conducted. 123 Other drugs such as various flavonoids 19, 124 bacitracin 125 or selenium, 126 by means of inhibition of protein disulfide isomerase, are other potential possibilities.
TF Inhibition in COVID-19
As previously commented, it is too early to know the true role of the over-expression of TF in COVID-19; likewise, the possible therapies derived from these findings are just beginning to be formulated. Inhibition of TF could be a therapeutic strategy in patients with COVID-19 who are initiating thrombotic coagulopathy, whose criteria would be of a clinical nature associated with an increase in D-dimer. ACE-I drugs due to their mechanism of action have been the object of study during the pandemic; although it was initially thought that they could be beneficial, 127 it has been shown that they do not intervene with the course of the disease, suggesting users of these medications should not discontinue or change their treatment. 128 Antithrombotic therapy in COVID-19 is based in low molecular weight heparins and aspirin; intermediate-dose anticoagulation and aspirin were each associated with a lower cumulative incidence of in-hospital death. 129 The impact of salicylate therapies on overexpressed TF in patients with COVID-19 remains to be demonstrated. Celecoxib, is a candidate for treatment of COVID-19, taking into account its effect as an anti-inflammatory medication 130 ; it would also be necessary to study the effect as an inhibitor of TF. Cyclosporine A might be a candidate in severe COVID-19 to prevent the cytokine storm (or hyperinflammation) and inactivate viral replication (117) disregarding its potential role in inhibiting TF and possible antiviral effect. 131 Pentoxifylline has also been proposed for the treatment of COVID-19 as a drug with antiviral, anti-inflammatory and bronchodilatory effects. 132 Colchicine recently associated with impact of on mortality in patients with COVID-19 133,134 ; In addition to its anti-inflammatory effect, it may also exhibit an inhibition of TF 135,136 and has a possible antiviral effect. 137
Vitamin D deficiency has been associated with risk for the development of COVID-19 and its deficiency must be corrected 138 ; its effect on immune dynamics is postulated, 139 however its effect as a TF inhibitor has yet to be demonstrated. Recent reviews speculate on the usefulness of antioxidants 140,141 and selenium. 126
Conclusion
The coagulopathy associated with COVID-19 appears to be multifactorial, involving inflammatory processes; endothelial and platelet dysfunction; the presence of antiphospholipid antibodies; and, because of the increase in AT2 in response to ACE-2 down-regulation, quite possibly TF overexpression. Different therapeutic targets have been derived from TF expression inhibition, which should be targeted by possible therapeutic candidates for the coagulopathy associated with COVID-19.
Footnotes
Acknowledgment
The authors thank Dr. David Ascher for the valuable review of article.
Author Contributions
All authors contributed to the preparation of the work and writing of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.