
Abstract
von Willebrand disease (vWD) is the most common inherited disorder of hemostasis and comprises a spectrum of heterogeneous subtypes. Significant advances have been made in understanding von Willebrand factor (vWF) gene mutations, resultant physiologic deficits in the vWF peptide, and their correlation to clinical presentation. Diagnostic tests for this disorder are complex, and interpretation requires a thorough understanding of the underlying pathophysiology by the practicing physician. The objective of this review is to summarize our current understanding of pathophysiology, laboratory investigations, and evolving treatment paradigm of vWD with the availability of recombinant von Willebrand factor.
Keywords
Introduction
The first case of von Willebrand disease (vWD) was described by Dr Eric von Willebrand in 1926. 1 The index case was a 13-year-old female who bled to her demise during her fourth menstrual period. Four of her 7 siblings were also affected. Dr von Willebrand noted several distinct characteristics of this bleeding disorder that contrasted it from hemophilia. First, both genders were affected. Second, there was prolongation of bleeding time. Third, phenotype was characterized mostly by mucocutaneous bleeding, as opposed to deep subcutaneous or joint bleeding, as noted in hemophilia. He called this disease “hereditary pseudohemophilia.” 1 The clinical characteristics described in 1926 hold true even till date and perfectly summarize this bleeding disorder; however, the diagnostic and management options have evolved considerably. In this review, we summarize our current understanding of von Willebrand factor (vWF) and disorders of hemostasis resulting from its deficiency.
Synthesis, Structure, Clearance, and Function of vWF Protein
The vWF is synthesized in endothelial cells and megakaryocytes, and it undergoes a series of complex posttranslational modifications. The gene for vWF is located on the short arm of chromosome 12. Translation of this gene results in the formation of a 2813-amino-acid-long pre-pro-vWF molecule, which consists of a 22-amino-acid-long “signal peptide,” 741 amino-acid-long propeptide, and a 2050-amino-acid-long mature subunit (Figure 1). 2,3 Following translation, the “pre-pro-vWF” is guided by the N-terminal signal peptide into endoplasmic reticulum, where signal peptide is cleaved, and dimerization occurs through tail–tail disulfide bond formation between creatine kinase (CK) domains of pro-vWF monomers. 4,5 The dimers are then transported to Golgi complex where multimerization occurs through head-to-head disulfide bond formation between D3 domains of adjacent molecules (Figure 1). 6,7 Further modification continues in the post-Golgi compartment and includes addition of ABO groups and glycosylation; changes that will affect vWF proteolysis by a disintegrin and metalloproteinase family protein with thrombospondin type 1 motif, member 13 (ADAMTS-13) and protect mature vWF from proteolytic clearance. 8 –10 The mature subunit that gets secreted into circulation possesses all the adhesive sites required for hemostatic function of vWF. These consist of D′-D3 domain that binds factor VIII, A1 domain that binds platelet GpIb receptor, A2 domain that serves a site for ADAMTS-13-mediated cleavage of vWF, A3 domain that binds exposed collagen, and C1 domain that binds platelet glycoprotein IIb/IIIa. 3,5,11 –16 The flanking domains, although not present on a mature vWF peptide, have important role in structural configuration of mature vWF multimers. The CK domain aids in the formation of disulfide bond between vWF monomers (tail-to-tail dimers), and D1-D2 domain promotes head-to-head oligomerization between D3 domains of vWF dimers to form large-molecular-weight vWF multimers (LMWMs) and ultralarge-molecular-weight multimers (ULMWMs), the most physiologically active forms of vWF. 5,6,7 The hemostatically less active, smaller multimers are secreted in a constitutive fashion by the endothelial cells and megakaryocytes; however, the more biologically active LMWMs and ULMWMs are preferentially targeted into storage organelles called Weibel–Palade bodies of endothelial cells and alpha granules of platelets and are secreted following endothelial or platelet activation. 17 –23
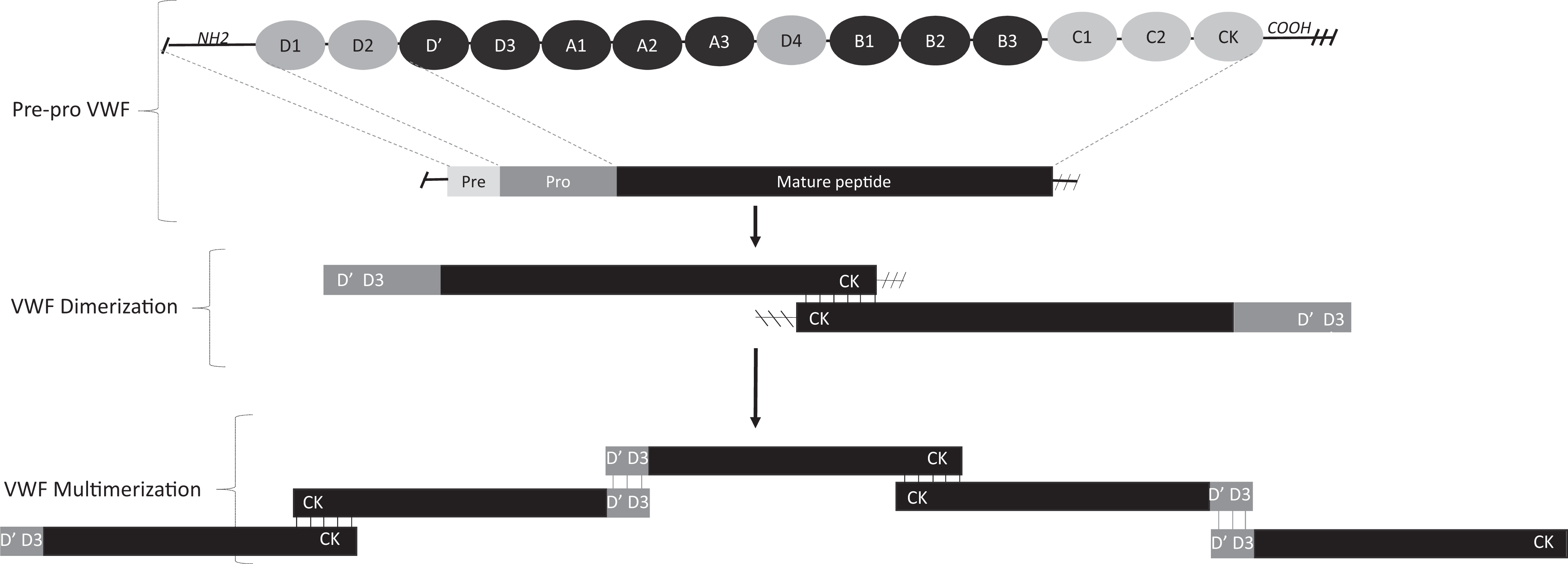
Synthesis and structure of von Willebrand factor (vWF). Translation of vWF gene results in the formation of a pre-pro-vWF molecule, which consists of a signal peptide, pro-peptide, and mature subunit. Dimerization occurs through tail–tail disulfide bond formation between creatine kinase (CK) domains of pro-vWF monomers, following the cleavage of N-terminal. Multimerization occurs through head-to-head disulfide bond formation. The CK domain aids in disulfide bond formation between vWF monomers (tail to tail dimers), and D1-D2 domain promotes head-to-head oligomerization between D′-D3 domains of adjacent vWF dimers (head-head multimers) to form large-molecular-weight multimers (LMWMs) and ultralarge-molecular-weight multimers (ULMWMs).
Clearance of vWF from plasma is not well understood; however, macrophages in liver and spleen are believed to internalize and clear circulating vWF. 24 Although, there is a wide variability in the half-life of endogenous vWF following desmopressin (DDAVP) challenge (range: 4-25 hours), the half-life of the plasma-derived therapeutic vWF is approximately 12 to 14 hours. 25,26 Several factors that influence such clearance have been outlined. Most prominent of these are the changes in glycosylation of vWF, addition of ABO groups 27 –29 , and point mutations in domains D3, D4, and A1. 30 –33 Although most patients with accelerated clearance have normal functional levels of vWF, point mutations in D3 have been commonly described to result in accelerated clearance of vWF and a pathologic phenotype mirroring type 1 or 2A vWD. The ADAMTS-13 has an important regulatory role in vWF physiology. As ULMWMs unfold under sheer stresses of circulation, A2 domain is exposed and serves as a site for attachment and cleavage by ADAMTS-13. The ADAMTS-13 cleaves ULMWMs into smaller, less hemostatically active multimers, such as intermediate-molecular-weight multimers and LMWMs. 10,24,34,30 Such proteolysis with ADAMTS-13 however has not been shown to accelerate the clearance of vWF. 34
Role in Hemostasis
Following endothelial injury, the stimulated endothelium and recruited platelets release the stored vWF. 19,20 The released nascent vWF then binds to the exposed collagen through A1 and A3 domains of vWF, resulting in vWF immobilization at the injury site. Once immobilized, vWF is subject to high sheer forces of circulation and undergoes conformational change exposing GpIb-binding site within its A1 domain. 31,32 The GpIb within the membranes of circulating platelets can then bind to the exposed GPIb-binding site on vWF, resulting in platelet adhesion. Adhered and activated platelets then roll on endothelium allowing GpIIb/IIIa conformational change and subsequent binding of platelet GpIIb/IIIa to vWF through its C1 domain, a slow but irreversible process, culminating in the formation of an initial platelet plug. 16,33 –36 The vWF also serves to bind and stabilize coagulation factor VIII (FVIII) through D′-D3 domains. 11 Activated FVIII can then activate factor X through the formation of “Xase complex” with FIX and calcium. This activation of coagulation cascade eventually results in the formation of a fibrin clot. 11,13,37 Thus, vWF plays an important physiologic role in platelet adhesion (through GpIb binding), platelet aggregation (though GpIIb/IIIa mediated binding) as well as in coagulation by prolonging the half-life of FVIII (Figure 2).
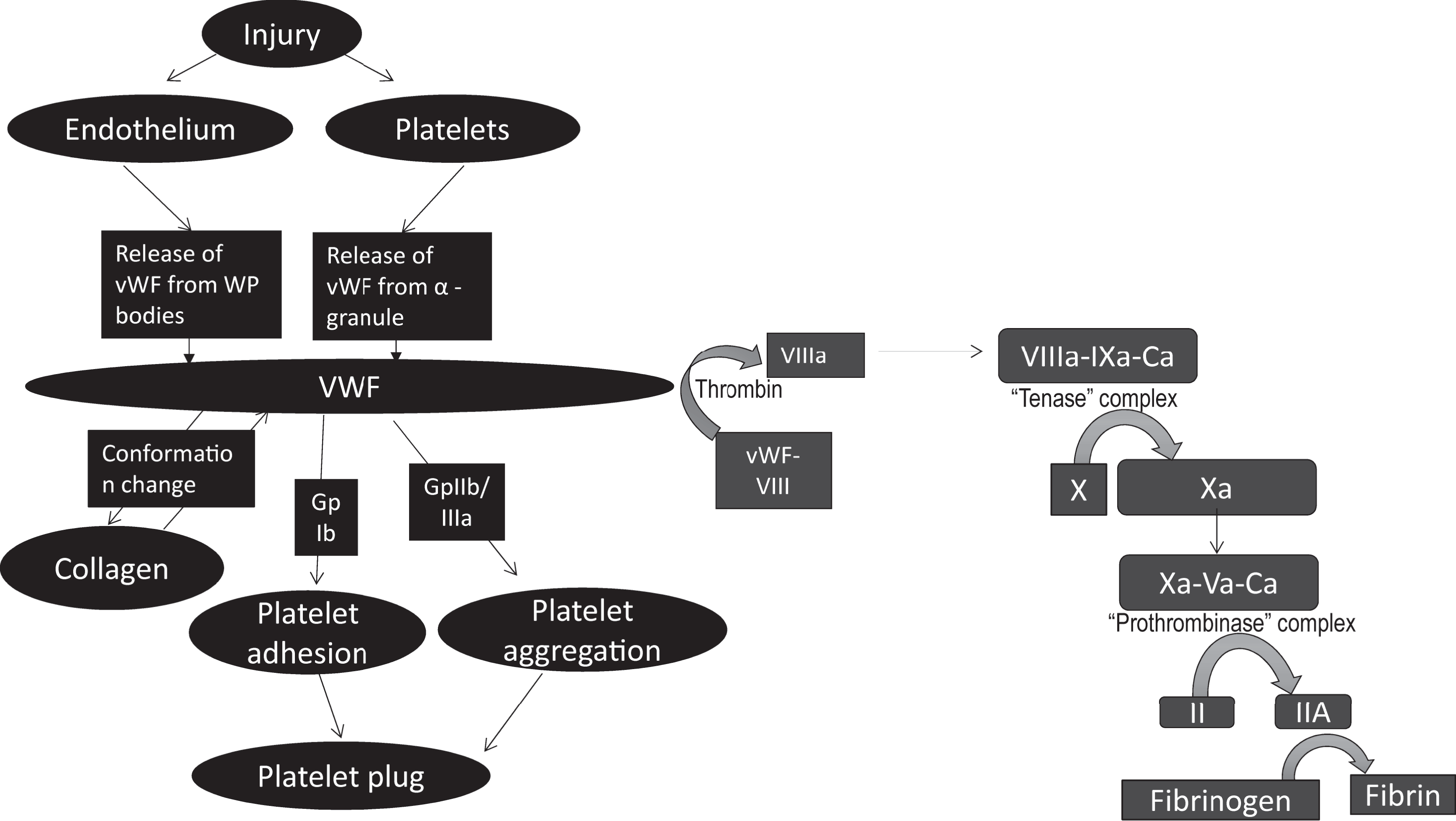
Function of vWF. At the injury site, stimulated endothelial cell and recruited platelets release stored vWF which binds to collagen through A3 domain, resulting in immobilization of vWF. Once immobilized, vWF is subject to high sheer forces of circulation and undergoes conformational change exposing GpIb binding site within A1 domain. Circulating platelets bind to exposed GpIb binding site on immobilized vWF, resulting in platelet adhesion. Adhered platelets then roll on endothelium until GpIIb/IIIa-mediated binding can occur resulting in platelet aggregation and formation of a platelet plug. The vWF also serves to bind and stabilize FVIII. Once FVIII is activated, it serves to activate Factor X through the formation of “Xase” complex with FIX and calcium. Activation of coagulation cascade eventually results in the formation of a fibrin clot.
von Willebrand Disease
The vWD is a disorder of hemostasis that results from deficient or defective plasma vWF. It is the most commonly inherited bleeding disorder, affecting 0.1% to 1% of the total population. 38 Due to incomplete penetrance and mild deficiency of vWD, some patients with vWD remain asymptomatic and are only diagnosed if testing is performed as a part of genetic screening if a relative has symptomatic disease. 39,40 Prevalence of symptomatic vWD is much lower, estimated at only 0.01% of the population. 38 Either gender can be affected, as the most common vWD variants undergo an autosomal inheritance pattern. Clinically, vWD presents with mucocutaneous or “platelet-type” bleeding, although “hemophilia-like” overlap presentation may be seen in some cases where FVIII levels are sufficiently low, such as in cases of types 2N and 3 vWD. Age of symptom onset is variable, but severe vWF deficiency, especially homozygous or doubly heterozygous variants of type 3 vWD, will manifest in childhood with joint bleeds on crawling, ambulating, or with minimal trauma, mirroring the clinical presentation of hemophilia. It is important to note that most patients with vWD have no or few symptoms only, with hemorrhage presenting only after trauma/surgery or mild spontaneous bleeding such as nose bleed or menorrhagia. The clinical presentation of various subtypes is further elaborated under descriptions of vWD subtypes below and in Table 1. Epistaxis, bleeding following dental extractions, easy skin bruising, menorrhagia, gingival bleeding, excessive posttraumatic bleeding, and postpartum bleeding are the common presenting manifestations of this disease. However, the less common variants types 2 and 3 vWD can present with life-threatening bleeding. 39,41 –53 Diagnosis and management of vWD rely heavily on accurate and objective personal and family history of bleeding. Several bleeding assessment tools have been developed and validated. One such well-recognized tool is the International Society of Thrombosis and Hemostasis (ISTH) bleeding assessment tool, which allows a quantitative scoring of bleeding symptoms. 54 According to the current classification of ISTH, vWD is categorized into 3 types:
Type 1 is characterized by a partial quantitative deficiency of essentially normal vWF.
Type 2 is characterized by qualitative defects in vWF.
Type 3 is characterized by an almost complete quantitative deficiency of vWF.
Clinical Characteristics of Various Subtypes of vWD.

Abbreviations: AD, autosomal dominant; ADAMTS-13, a disintegrin and metalloproteinase family protein with thrombospondin type 1 motif, member 13; AR, autosomal recessive; CK, creatine kinase; vWD, von Willebrand disease; vWF, von Willebrand factor.
Clinical presentation, common mutations, and laboratory characteristics of various types of vWD are listed in Tables 1 and 2.
Laboratory Characteristics of vWD Subtypes.

Abbreviations: IMWMs, intermediate molecular weight multimers; LMWM, large-molecular-weight multimers; PTT, partial thromboplastin time; RIPA, low-dose ristocetin-induced platelet aggregation; vWD, von Willebrand disease; vWFpp, von Willebrand factor pro-peptide; vWFpp/vWF:Ag, vWF propeptide/vWF antigen ratio; vWF:Ag, von Willebrand factor antigen; vWF:CB, von Willebrand factor collagen binding; vWF:FVIIIB, von Willebrand factor and FVIII binding; vWF:RCo, von Willebrand factor:ristocetin cofactor activity; vWF: RCo/vWF:Ag, vWF ristocetin cofactor activity/vWF antigen ratio.
Type 1 vWD is the most common subtype, accounting for approximately 70% of vWD cases and follows an autosomal-dominant (AD) inheritance pattern. 53 In type 1 vWD, there is a decrease in the concentration of functionally normal vWF either due to decreased synthesis/secretion 39 or due to increased clearance (also known as type 1C). 42 Y1584C is the most commonly implicated point mutation in type 1 vWD, although several different mutation including frameshift mutations, deletions, and other point mutations have been characterized. 39 Clinically, patients may be asymptomatic or present with mild-moderate mucocutaneous bleeding.
Type 2 vWD accounts for 25% cases and is characterized by qualitative deficiency of vWF activity. 52 It is further subclassified into 4 subtypes: 2A, 2B, 2M, and 2N. Type 2 vWD should be suspected when there is evidence of functional discordance in vWF activity and antigen levels (vWF:ristocetin cofactor activity [RCo]/vWF:Ag ratio of <0.6). Bleeding symptoms are more severe in type 2 vWD than in type 1 vWD. Clinically, most patients present with platelet-type mucocutaneous bleeding; however, in subtypes with markedly decreased FVIII levels, “hemophilia-type” joint and deep subcutaneous phenotype can dominate, such as type 2N (Table 1).
Type 2A vWD is inherited as an AD trait and is characterized by a decrease in high- and intermediate-weight multimers. 41 This subtype arises from missense mutations that lead to a net fewer GpIb binding sites on vWF and thus decreased effectiveness in platelet clot formation. 43 Such deficiency in vWF function can result through various mechanisms, such as defects in dimer or multimer assembly, impaired secretion, increased intracellular retention, or enhanced proteolysis of vWF. Table 1 lists the common mutation domains and resultant functional deficits in mature vWF seen in type 2A vWD. 43 –46 Clinically, moderate to severe mucocutaneous bleeding is seen.
Type 2B vWD also has an AD inheritance and is characterized by loss of large-molecular-weight multimers; but the mechanisms for such change are quite distinct from type 2A. Gain of function mutations in or around the A1 domain (exon 28) results in an increased affinity of GpIb binding site on vWF for the platelet GpIb receptor, leading to spontaneous binding in circulation. 47,48 The large multimers have higher number of GpIb binding sites and are thought to be rapidly cleared from plasma, along with bound platelets, thus resulting in characteristic thrombocytopenia and lack of LMWMs in patient’s plasma. This characteristic is also demonstrated in vitro when increased binding is seen with low concentrations of ristocetin on ristocetin-induced platelet aggregation (RIPA) testing. Clinical phenotype usually is of moderate to severe mucocutaneous bleeding.
Type 2M vWD is a less common variant of type 2 vWD that has an AD inheritance and is characterized by a qualitative defect that is not due to decreased high molecular weight multimers (HMWMs). Mutations implicated in this phenotype impair vWF-platelet (domain A1-GpIb) 49 and vWF-collagen (domain A3-collagen) interactions. 50 On laboratory testing, there will be normal multimer pattern and reduced RIPA sensitivity. It is important to note that reduced vWF collagen binding (vWF:CB) may be witnessed, especially with mutations in vWF domains A1 or A3 that are important for vWF-collagen interaction. This subtype commonly presents with moderate to severe mucocutaneous bleeding.
Type 2N vWD is a rare variant with an autosomal-recessive inheritance characterized by a defect in vWF which interferes with its binding to FVIII. Mutations in domain D′-D3 (exons 18-21) which result in reduced affinity of vWF to FVIII have been implicated. 55 In laboratory test results, a characteristic decline in FVIII levels relative to vWF levels (reduced FVIII/vWF:Ag ratio) was observed, with a prolonged partial thromboplastic time (PTT) and decreased vWF:FVIII binding, despite a normal multimer profile. Clinically, both platelet-type and hemophilia-like bleeding are seen. This subtype can mimic hemophilia A, thus in addition to FVIII levels, qualitative and quantitative vWF assays, including vWF:FVIII binding assays, are required to establish a diagnosis.
Type 3 vWD is a rare subtype, accounting for less than 5% of vWD cases and is characterized by virtual absence of vWF. 52 Mutations associated with type 3 vWD are found throughout the coding region of vWF gene (exons 2-52), with 80% of cases having 2 null alleles. 56,57,58 A rare instance is mutations in D1-D2 domains, which prevents dimer and multimer formation and thus intracellular retention of all synthesized vWF. 59 In laboratory test results, virtual absence of vWF multimers, undetectable vWF antigen and vWF:RCo, prolonged PTT, and markedly reduced FVIII levels (<10IU/L)were observed. Clinically, there is severe mucocutaneous as well as joint and subcutaneous tissue bleeding.
In addition to inherited deficiency, bleeding disorders from acquired vWF deficiency (acquired von Willebrand syndrome (AVWS)) have been reported in association with a variety of underlying disorders. Clinical presentation is similar to inherited vWD but without a personal of family history of bleeding tendencies. Underlying mechanisms may include reduced synthesis, antibody-mediated functional interference or accelerated clearance, complex formation with plasma proteins, adsorption onto surface of circulating cells or platelets, and heightened lysis under increased sheer stress as seen in aortic stenosis (Table 3). 60 –62

Diagnostic Evaluation of Suspected vWD
A thorough understanding of vWF physiology and provision of a standardized laboratory are essential in establishing a diagnosis of vWD. Due to assay variability, limited sensitivity of certain assays, and preanalytic issues with sample handling, diagnosis of vWD may be missed in a significant proportion of individuals with the disease. It is thus recommended that at least 2 sets of tests using fresh samples be collected to confirm or refute the diagnosis of vWD. Laboratory investigations can be subdivided into following 3 broad categories: Preliminary tests: These tests are not recommended for screening or diagnosis of vWD but invariably serve as starting points for workup following a hematology referral. These include complete blood count (CBC), prothrombin time, and PTT. The CBC profile may be normal but may show microcytic anemia due to iron deficiency from ongoing blood loss and thrombocytopenia in type 2B vWD. The PTT is normal in majority of cases but may be prolonged if FVIII levels are reduced to <30 to 40 IU/dL (severe type 1, type 2N, and type 3). Tests to establish diagnosis: vWF Antigen levels (vWF:Ag) determine the quantity of vWF protein in plasma. Enzyme-linked immunosorbent assay (ELISA) or latex immunoassay (LIA) method is used. Normal range is 50 to 200 IU/dL.
57,63
Ristocetin cofactor activity assay (vWF:RCo) determines the capacity of vWF to agglutinate exogenous platelets in the presence of ristocetin. The rate of ristocetin agglutination is related to the concentration of functionally normal vWF. It is noteworthy that circulating functionally and structurally normal vWF in plasma does not bind platelet GpIb unless conformational change is brought in vWF by binding of vWF to subendothelium, exposing GpIb binding sites of vWF. The antibiotic ristocetin is thought to mimic the subendothelium to bind and immobilize vWF, thus causing conformational change and aiding the binding of vWF to platelets, resulting in platelet agglutination and subsequent aggregation. Aggregometer or LIA method is used. Normal range is 50 to 200 IU/dL.
63,51
vWF:RCo/VWF:Ag ratio assesses the ratio of vWF activity to antigen in plasma. A ratio of <0.6 is indicative of a qualitative vWF deficiency. A value of <0.6 is seen in all type 2 vWD subtypes.
51,64
Factor VIII coagulant assay (FVIII:C) measures the plasma concentration of FVIII. To assay the ability of plasma to shorten PTT of FVIII-deficient plasma. Normal range is 50 to 200 IU/dL.
64
Tests to classify vWD subtypes: vWF multimer analysis is a qualitative assay that determines the size distribution of vWF multimers. Sodium dodecyl sulfate protein electrophoresis method is used. This test can be described as a 2-step process. First, the plasma is subjected to electrophoresis, and vWF multimers migrate based on the charge and size, with the smallest multimers migrating farthest. Second, immune detection of multimers is undertaken by employing radio-labeled antibodies. Results are interpreted either through visual inspection or by densitometry (Figure 3A).
65
Low-dose RIPA: measures the affinity with which vWF binds to platelet GpIb receptor, by limiting the quantity of ristocetin. Platelet-rich patient plasma (provides both platelets and vWF) is mixed with sequentially lower concentrations of ristocetin (range: 0.4-1.2 mg/mL), and the presence/absence of platelet aggregation at each concentration is assessed using aggregometry (Figure 3B). Ristocetin concentration <0.6 mg/mL fails to cause vWF-GpIb binding in samples from normal patients but is able to bring about aggregation in patients with type 2B vWD, which is characterized by increased affinity of vWF for GpIb receptor.
48
The RIPA assumes that patient’s platelets are quantitatively and qualitatively normal; however, if abnormal or impaired platelet GpIb (decreased number or affinity) coexists, platelets will fail to aggregate, despite the presence of functionally normal vWF. Another important consideration is an entity called pseudo-vWD or platelet-vWD, which is characterized by mutations in platelet GpIb that increase the affinity of mutated platelet GpIb for normal vWF. Type 2B vWD and platelet-type vWD can be distinguished by employing a modified RIPA approach.
66
In this approach, when RIPA is performed using patient’s plasma (vWF) and exogenous platelets, normal aggregation pattern will be seen. However, when RIPA is performed using patient’s platelets and exogenous plasma (vWF), increased aggregation at low concentration becomes evident, thus identifying abnormality in platelets rather than vWF. vWF-FVIII binding assay (vWF/FVIII:B) measures the ability of vWF to bind exogenous FVIII. ELISA is used. For this test, patient and control plasma are applied to plates coated with anti-vWF antibodies. These plates are then incubated with calcium chloride to remove all endogenous FVIII. Exogenous recombinant FVIII is then added followed by tagged FVIII antibodies. Colorimetric assay is performed, and the curve generated from patient’s plasma is plotted against a standard or control curve. Defective vWF/FVIII:B is seen in type 2N vWD (Figure 3C).
67
vWF-collagen binding assay (vWF:CB) measures the affinity of vWF for collagen. Patient and control plasma are applied to collagen-coated ELISA plates, followed by immune detection of collagen-bound vWF using antihuman vWF antibodies. Defective CB is seen in vWD 2M, with mutations in CB domains A1 or A3 of vWF. These variants may have normal vWF:RCo and thus escape diagnosis, unless vWF:CB testing is performed.
31,32,68
Optimal CB of vWF is dependent on the quantity of HMWMs that readily bind collagen, thus vWF:CB testing is useful in distinguishing type 1 vWD from types 2A, 2B, and 2M.
68
vWF propeptide/vWF antigen ratio (vWFpp/vWF:Ag) measures the ratio of vWF pro-peptide levels and mature vWF in plasma. An increased ratio, implying increased vWFpp concentration in comparison to mature vWF antigen in a patient with vWD, is indicative of adequate synthesis and secretion of vWF but an increased clearance of vWF as the likely underlying etiology of vWF deficiency.
59
The DDAVP challenge testing (DDAVP responsiveness:) is advisable after a diagnosis of vWD is established, especially in mild-moderate types 1 and 2 vWD. vWF antigen and activity, along with FVIII levels are determined before and at 1, 2, and 4 hours after DDAVP administration (0.3 mcg/kg IV). Response is considered adequate if a 2- to 4-fold increase in vWF:Ag and vWF:RCo and at least a 30% increase in FVIII levels are seen that last for 6 to 12 hours. Adequate response is seen in 90% of patients with type 1 vWD, and in these patients, DDAVP alone will be adequate for prophylactic management for bleeding associated with dental procedures/minor surgeries as well as treatment of menorrhagia or epistaxis. On the other hand, an adequate initial response but lasting less than 4 hours is indicative of increased vWFclearance. The DDAVP alone may not achieve adequate hemostasis in these patients. It should be noted that DDAVP responsiveness testing is contraindicated in type 2B vWD, as it may promote binding of secreted vWF to platelets in vivo and worsen thrombocytopenia.
69,70
Genotyping: Although several point mutations in vWF gene have been identified, DNA sequencing for detection of vWF gene mutations is not commonly employed for diagnosis and is not widely available. This is attributed to the complex genetic and phenotypic variability seen in vWD, frequent single-nucleotide polymorphisms in vWF gene, large size of vWF gene, and presence of a homologous pseudogene at chromosome 22. Mutation analysis can be utilized in detecting mutations associated with most type 2 vWD subtypes because these mutations cluster in a geographically narrow area of the gene (exons 18-32).
71,72
Diagnostic considerations in AWVS: In addition to the above testing, a vWF:RCo mixing study should be considered in patients with clinical suspicion of AWVS. Reduced vWF:RCo noted in an adult patient with bleeding and no notable personal or family history of bleeding tendencies should arouse suspicion for AVWS. To screen for AVWS, a vWF:RCo mixing study should be performed next, using control plasma. Failure to normalize vWF:RCo on addition of control plasma indicates the presence of an inhibitor. Such inhibitory autoantibodies can then be detected using ELISA.
61,62
Note that autoantibodies to nonfunctional domains, which may accelerate vWF clearance in vivo but confer no functional interference, will be missed on such functional testing. Findings of various laboratory tests for different subtypes of vWD are summarized in Table 2.
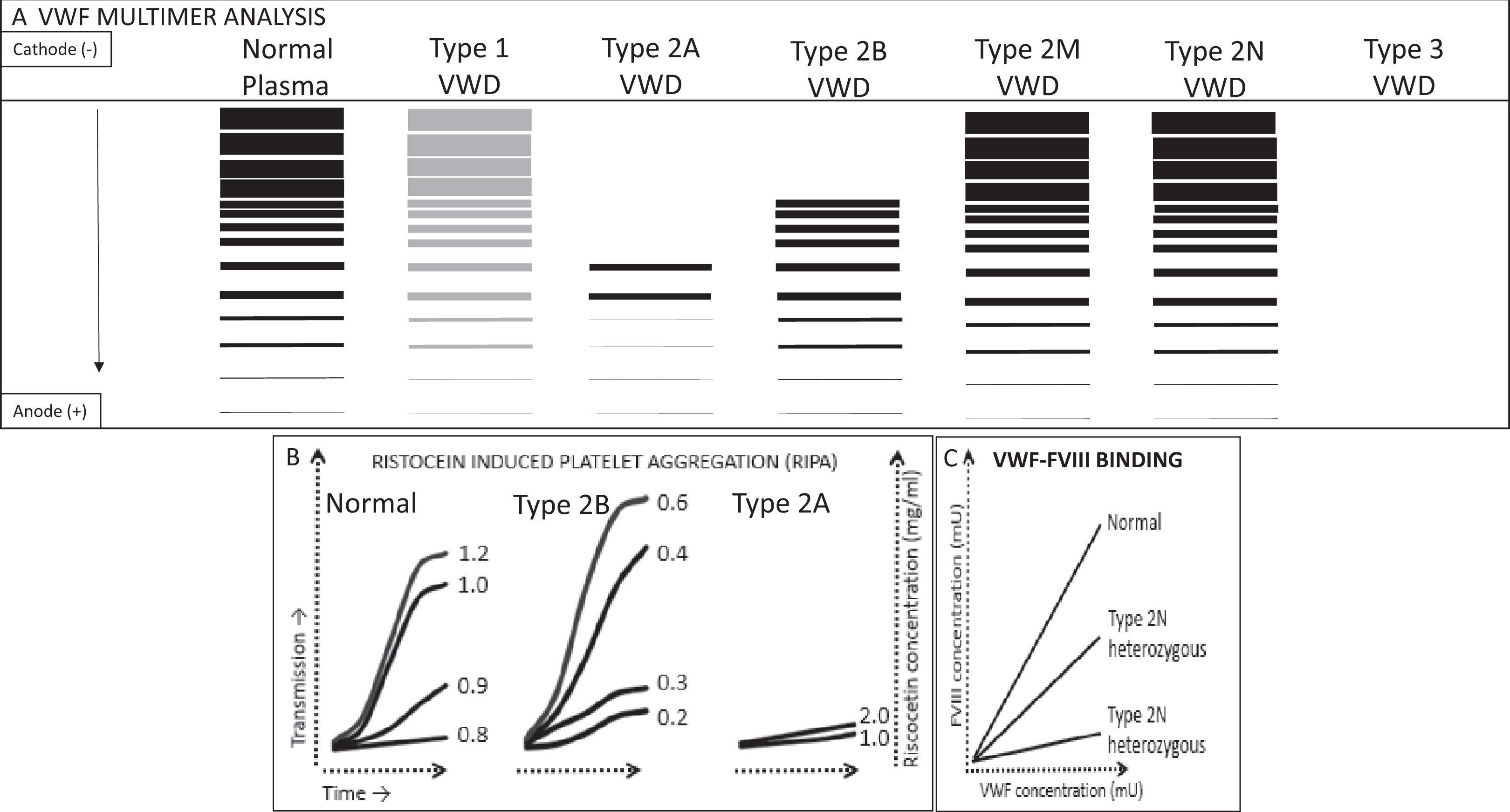
Laboratory tests in von Willebrand disease (vWD). A, vWF multimer analysis on protein electrophoresis, the vWF multimers migrate based on their charge and size, with lowest molecular weight multimers migrating the farthest. In type 1 vWD, all multimers are present, albeit in reduced concentration. In type 2A vWD, large- and intermediate-molecular-weight multimers are absent. In type 2B, Large-molecular-weight multimers are absent. In types 2M and 2N, full spectrum of multimers is seen, whereas in type 3 there is an almost complete absence of vWF. B, Shows platelet aggregation curve in the presence of low-dose ristocetin (ristocetin-induced platelet aggregation). In normal plasma, no aggregation occurs when the ristocetin concentration is below 0.8 mg/mL. In type 2B vWD (or platelet type vWD), aggregation occurs at much lower ristocetin concentrations, whereas in type 2A vWD, aggregation fails to occur even with much higher ristocetin concentration. C, Curves generated by colorimetric assay for FVIII and vWF to assess vWF-FVIII binding. vWF:FVIII binding is expressed in percentage compared to the standard curve (normal plasma). In type 2N vWD, vWF fails to bind to FVIII and thus lower bound FVIII percentage is seen.
Overview and Summary of Management Strategies for vWD
As a general rule, a thorough personal and family history, aspirin or nonsteroidal anti-inflammatory drug use, and history of antiplatelet therapy should be obtained for all patients being evaluated for disorders of hemostasis. The following treatment options can be employed in treating a bleeding patient with vWD
64
: Local measures: Minor bleeding from nasal or oral bleeding may be managed with prolonged local pressure to the injury site or with the use of certain topical hemostatic agents (Figure 2). Several topical agents have been employed including topical human thrombin or fibrin sealants, micronized collagen strips, and so on.
64,73
The use of bovine products should be prohibited because an increased risk of autoantibody formation to factor V has been reported with the use of such products.
73
Pharmacologic agents for indirect hemostatic effect: Fibrinolysis inhibitors: Epsilon aminocaproic acid inhibitor and tranexamic acid act by preventing dissolution of hemostatic plug, especially in mucosal bleeding. Bleeding with dental procedures and menorrhagia seen with mild type 1 vWD can be successfully treated with such agents.
74
Hormonal treatments: Estrogen–progesterone-combined oral contraceptives (OCPs) can be used successfully for treatment of menorrhagia in premenopausal women. Mechanisms for this effect are not entirely clear. The OCPs are thought to induce changes in the endometrium, making it less likely to bleed; however, there is also in vitro data showing that estrogens can increase the synthesis of vWF.
75
Therapies that directly increase vWF: Desmopressin: DDAVP, a synthetic analogue of human antidiuretic hormone, promotes the release of stored vWF from endothelial cells into circulation. The DDAVP responsiveness should be tested in all patients with type 1 and type 2A vWD. Optimal response occurs in the majority of patients with types 1 and 2A disease.
69,70
The DDAVP can be administered either intravenously or intranasally at doses of 150 to 300 mcg. Shortcomings of this therapy include development of tachyphylaxis as endothelium is depleted of stored vWF, hyponatremia, vasodilatation, and hypotension. The DDAVP is generally contraindicated in type 2B vWD and ineffective in type 3 vWD-related bleeding. Responsiveness to DDAVP is variable in vWD subtypes 2M and 2N. Some patients with these subtypes may benefit from DDAVP. The DDAVP responsiveness should thus be tested at baseline in patients with vWD subtypes 2M and 2N.
69,70
vWF/FVIII concentrates: Patients with bleeding associated with severe vWD (all types) and those with mild-moderate vWD requiring major surgeries require replacement therapy. Three plasma-derived intermediate purity FVIII/vWF concentrates are currently available in the United States: Humate-P, Alphanate, and Wilate.
76
–78
High-purity vWF concentrates are available in Europe; however, since most patients with vWF are also deficient in FVIII, intermediate purity concentrate replacement is preferred. The quantity of vWF:RCo relative to FVIII:C varies by product. Replacement is given to maintain vWF:RCo and FVIII levels at a peak of 100 IU/dL and a trough of 50IU/dL, until hemostasis.
64
Usual loading dose is 40 to 60 U/kg for mild-moderate type 1 or type 2 and type 3 vWD with minor bleeding; 50 to 75 U/kg for severe types 1, 2, and 3 vWD. In patients with major bleeding, maintenance doses are given every 8 to 12 hours for up to 7 days. Recombinant vWF: A recombinant human vWF was approved by US Food and Drug Administration in December 2015 for on-demand use in adult patients with severe bleeding. In a phase 3 trial “excellent” hemostasis and treatment success (bleeding score of <2.5) was achieved in 97% and 100% patients, respectively.
79
Management of this disease is a landmark achievement, considering the issues with the transfer of the large vWF gene. Given its excellent efficacy and safety profile, the use of this product is expected to continue to increase. Special issues in AWVS: Diagnosis and management of the underlying disorder is of utmost importance in the management of AVWS. In addition to the above approaches for treatment of WVD, a bleeding patient with AVWS may benefit from other interventions that target the inhibitor, including IVIG, plasma exchange, and immune suppressants or address the underlying cause, such as treatment of underlying severe hypothyroidism or associated malignancy. Recombinant FVII may be helpful in bypassing the inhibitor in emergency situations.
60
–62
Footnotes
Authors’ Note
Arjun Swami is now affiliated with St. George’s University, Grenada. Varinder Kaur is now affiliated with University of Virginia, Charlottesville, VA, USA.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.