
Abstract
Atrial fibrillation (AF) is a significant risk factor for stroke and peripheral thromboembolic events (TEs). Preventing blood clots in the heart to reduce stroke and TE risk is a key goal of AF therapy. Traditional stroke risk assessment tools for patients with nonvalvular AF include the CHADS2 and CHA(2)DS(2)-VASc scores, while long-term outcome data with the newer direct oral anticoagulants (DOACs) are emerging. The goals of this review were to assess traditional therapies and existing treatment guidelines and to discuss key pharmacologic properties of the DOACS, noting how these may benefit at-risk patients with AF. This narrative review was developed on the basis of the authors’ clinical knowledge, extensive reading of the literature, and broad pharmacy experience in the management of patients with AF. Limitations of oral vitamin K antagonists (VKAs) include slow onset of action, the need for regular monitoring of their anticoagulation effect, significant food and drug interactions, and unpredictable dose–response properties. Key clinical trial data led to the approvals of apixaban, dabigatran etexilate, edoxaban, and rivaroxaban in the United States to reduce the risk of stroke and systemic embolism in patients with nonvalvular AF. With predictable pharmacologic properties and limited drug and/or dietary interactions, the DOACs offer several benefits over traditional oral anticoagulation therapy with VKA. However, they have limitations, including the absence of immediate reversal agents and limited options for monitoring their anticoagulation effects in clinical practice. As experience with the use of DOACs grows, optimized treatment regimens and improved patient care are expected.
Keywords
Introduction
Overview of Atrial Fibrillation
Atrial fibrillation (AF) is the most common form of cardiac arrhythmia 1 and is a significant risk factor for strokes and peripheral thromboembolic events (TEs). In addition, AF can lead to negative effects on cardiac function and output. 2 Well-known risk factors for AF include older age, male sex, hypertension, valvular disease, left ventricular dysfunction, obesity, and alcohol consumption. 3 In the Framingham Heart Study, the cumulative incidence of new cases of AF at the 22-year follow-up assessment was 21.5 per 1000 men and 17.1 per 1000 women. 4 It should be noted that the term “nonvalvular AF” (NVAF) is used to describe cases where the disturbance in the rhythm of the atria occurs in the absence of a prosthetic heart valve or mitral stenosis associated with a history of rheumatic fever. 5 For patients with AF, the 3 major goals for the management of AF are (1) rate control, (2) prevention of thromboembolism, and (3) the correction of rhythm disturbances. 6
Pathophysiology of Cardioembolism in AF
The lack of effective atrial contractions due to AF leads to blood stasis in the atria. This poor circulation can result in the formation of blood clots. Once formed, especially if they arise in the left atrial appendage, these clots can be ejected into the arterial circulation and lead to TEs in the brain and elsewhere (systemic embolisms). 7
Stroke Risk Assessment Tools in AF
Patients with AF are at increased risk of a stroke, but the relative risk (RR) for each individual varies depending on a number of patient factors including age, sex, and the presence of 1 or more well-known comorbid conditions such as hypertension, diabetes, prior TEs, and so on. These have been formally combined into the CHADS2 risk assessment scheme, which is calculated by assigning 1 point each for Congestive heart failure, Hypertension, Age ≥75 years, and Diabetes mellitus, with 2 points added for a prior history of Stroke or transient ischemic attack. 8 The CHADS2 scheme has performed well at the higher risk range but has not been as accurate for stratifying patients in the lower risk ranges. As a result, the scheme was updated by incorporating the Birmingham 2009 schema, resulting in the CHA(2)DS(2)-VASc (Congestive heart failure/left ventricular dysfunction, Hypertension, Age ≥75 years, Diabetes mellitus, Stroke/TIA/TE, Vascular disease, Age 65-74 years, Sex category), which demonstrated a positive correlation between TE rates and higher scores (P value for trend = .003). 9 This scoring algorithm was highly predictive of outcomes at the lower end of the spectrum, with 0 (0%) of 103 of patients with a score of 0 experiencing a stroke or other TE at 1 year postevaluation. 9 Although apparently also predictive at the higher end, where 2 (20%) of 10 of patients with scores of 8 or 9 experienced stroke or other TE, 9 this percentage is based on a very limited number of participants (2 of 10). Therefore, these results should be interpreted with caution.
Assessment Tools for Estimating Bleeding Risk Associated With Warfarin Therapy
To develop a model to quantify bleeding risk among elderly patients with AF, a composite registry of data and outcomes for 3791 Medicare beneficiaries was analyzed. 10 HEMORR2HAGES, an extension of existing schemes, was scored according to the following algorithm (2 points for a Rebleeding risk [ie, prior bleed] and 1 point for each of the other risk factors: Hepatic or renal disease, Ethanol abuse, Malignancy, Older [age >75 years], Reduced platelet count or function, Hypertension [uncontrolled], Anemia, Genetic factors, Excessive fall risk, and Stroke). 10 At the time this scheme was developed (2006), it was considered the best tool available for scoring bleed risk among patients on warfarin. 10
The use of anticoagulants in patients with AF can lead to increased risk of major bleeding events, including intracranial bleeding, decreases in hemoglobin >2 g/L, transfusion, and hospitalization. The HAS-BLED score (Hypertension, Abnormal renal/liver function, Stroke, Bleeding history or predisposition, Labile international normalized ratio, Elderly [>65 years], Drugs/alcohol concomitantly) was developed as a simple tool to assess bleeding risk in patients with AF. 11 Continued analysis of data from the Anticoagulation and Risk Factors in Atrial Fibrillation (ATRIA) off-warfarin cohort identified a number of factors (age, female sex, diabetes mellitus, chronic heart failure, hypertension, proteinuria, an estimated glomerular filtration rate of <45, and end-stage renal disease) as predictive factors, which when scored according to the algorithm constituted a good indicator of overall bleeding event risk. 12
Society Guidelines/Recommendations for Stroke Prophylaxis in Patients With AF
The American College of Cardiology/American Heart Association (ACC/AHA) Task Force on Practice Guidelines, in collaboration with the Heart Rhythm Society, recently issued updated recommendations for the prevention of TE in patients with AF. With regard to the appropriate selection of antithrombotic therapy for an individual patient, these guidelines recommend consideration of (1) the risk of thromboembolism irrespective of whether the AF pattern is paroxysmal, persistent, or permanent; (2) the patient’s preferences after discussion of the RRs associated with stroke and bleeding events; and (3) the overall risk of TE. Assessment of individual patient risk of stroke should include the use of the CHA(2)DS(2)-VASc score. 13
The 2014 ACC/AHA task force treatment guidelines also include the following recommendations: (1) for patients with mechanical heart valves, warfarin is the recommended agent, with target international normalized ratio (INR) intensity established according to valve location and prosthesis type; (2) in patients with a history of stroke, transient ischemic attack (TIA), or a CHA(2)DS(2)-VASc score ≥2, oral anticoagulation with warfarin, dabigatran, rivaroxaban, or apixaban is recommended; (3) patients on warfarin should have their INR assessed at least weekly during the initiation phase, followed by monthly monitoring once the patient is stable; (4) if a therapeutic INR is not maintained on warfarin, then use of a direct thrombin or factor Xa (FXa) inhibitor is recommended; (5) the clinical need for anticoagulation should be reassessed at regular intervals; and (6) antithrombotic therapy is recommended for patients with atrial flutter according to the same risk profile used for AF. 13
The 2012 American College of Chest Physicians (ACCP) issued extensive recommendations for the treatment of patients with nonrheumatic AF, stratified by stroke risk. 14 For patients at low risk (CHADS2 score = 0), the recommendation is no antithrombotic therapy. For patients who select antithrombotic therapy, the recommendation is for aspirin monotherapy rather than oral anticoagulation or combination therapy with aspirin and clopidogrel. 14 For patients who are at intermediate risk (CHADS2 score = 1), the recommendation is oral anticoagulation rather than no therapy, aspirin, or a combination of aspirin and clopidogrel, 14 and for patients who are at high risk of stroke (CHADS2 score ≥ 2), oral anticoagulation is recommended rather than no therapy, aspirin, or combination therapy with aspirin and clopidogrel. 14
Brief Review of Clinical Data for Traditional Therapies
The anticoagulant effects of warfarin have been extensively studied in a number of patient populations for the prevention of stroke in AF, and the relative benefits and risks of this mainstay therapy are well understood. For a brief overview of the major studies with this agent, see Table 1. Results from a few of the pivotal trials with warfarin are presented below.
Anticoagulation With Warfarin Versus Placebo.

Abbreviations: SPAF, Stroke Prevention in Atrial Fibrillation; AFASAK, Atrial Fibrillation, Aspirin, Anticoagulation; BAATAF, Boston Area Anticoagulation Trial for Atrial Fibrillation; CAFA, Canadian Atrial Fibrillation Anticoagulation; CI, confidence interval; EAFT, European Atrial Fibrillation Trial; GI, gastrointestinal; HR, hazard ratio; ND, not determined; NSD, no significant difference; RR, relative risk; SPINAF, Stroke Prevention in Nonrheumatic Atrial Fibrillation.
The Stroke Prevention in Atrial Fibrillation (SPAF-1) study was a multicenter, randomized trial with 1330 patients with AF. This study compared aspirin or warfarin with placebo for the prevention of ischemic stroke and systemic embolism. 15 The rates of ischemic stroke and systemic embolism were reduced 67% (P = .01) by warfarin and 42% (P = .02) by aspirin 325 mg daily. 15 The rates of significant bleeding events were 1.5%, 1.4%, and 1.6% per year (warfarin, aspirin, and placebo, respectively). 15
The combination of aspirin plus clopidogrel, or oral anticoagulation (vitamin K antagonist [VKA]) therapy, was investigated for the prevention of vascular events in 2 randomized trials. In the ACTIVE-W (Atrial fibrillation clopidogrel trial with irbesartan for prevention of vascular events) trial, patients with AF plus one or more risk factors for stroke were randomly allocated to receive oral anticoagulation therapy (target INR of 2.0-3.0; n = 3371) or clopidogrel (75 mg/d) plus aspirin (75-100 mg/d recommended; n = 3335). Patients on oral anticoagulation therapy experienced 165 primary events (3.93% annual risk), and patients taking clopidogrel plus aspirin had 234 primary events (5.60% annual risk; RR: 1.44; 95% confidence interval [CI]: 1.18-1.76; P = .0003). Major bleeding occurred at annual rates of 2.21% in patients receiving warfarin and 2.42% in patients receiving clopidogrel plus aspirin (RR: 1.10; 95% CI: 0.83-1.45; P = .53). This trial was terminated early due to clear superiority of oral anticoagulation therapy. 21 In the ACTIVE-A (Atrial fibrillation clopidogrel trial with irbesartan for prevention of vascular events)trial, 7554 patients with AF, who were at increased risk of stroke and not good candidates for VKA therapy, were randomly assigned to receive clopidogrel (75 mg) or placebo, once daily, in addition to aspirin. After a median follow-up of 3.6 years, major vascular events occurred in 832 patients (6.8%/year) on clopidogrel and in 924 patients (7.6%/year) on placebo (RR with clopidogrel 0.89; 95% CI: 0.81-0.98; P = .01). 22 Major bleeding occurred in 2.0% of patients receiving clopidogrel and in 1.3% of placebo patients (RR: 1.57; 95% CI: 1.29-1.92; P < .001).
The limitations of VKAs are well established and include the need for bridging upon initiation and when medical/surgical interventions are necessary, their slow onset of action, the need for regular monitoring of anticoagulation effects, a significant number of known drug–drug interactions, known interactions with food affecting pharmacokinetic (PK) and pharmacodynamic (PD) parameters, and their unpredictable dose-response properties. As a result of these limitations, an unmet medical need existed for alternative anticoagulants with PK and PD properties that would address these concerns while providing effective anticoagulation. Over the past several years, continued efforts in research and development have led to the approval for clinical use of 3 new agents, and details of their PK and PD properties, along with key results from recent pivotal clinical trials, are discussed below. General indications and dosing recommendations for each agent can be found in Table 2.
Indications and Dosing for Approved DOACs.
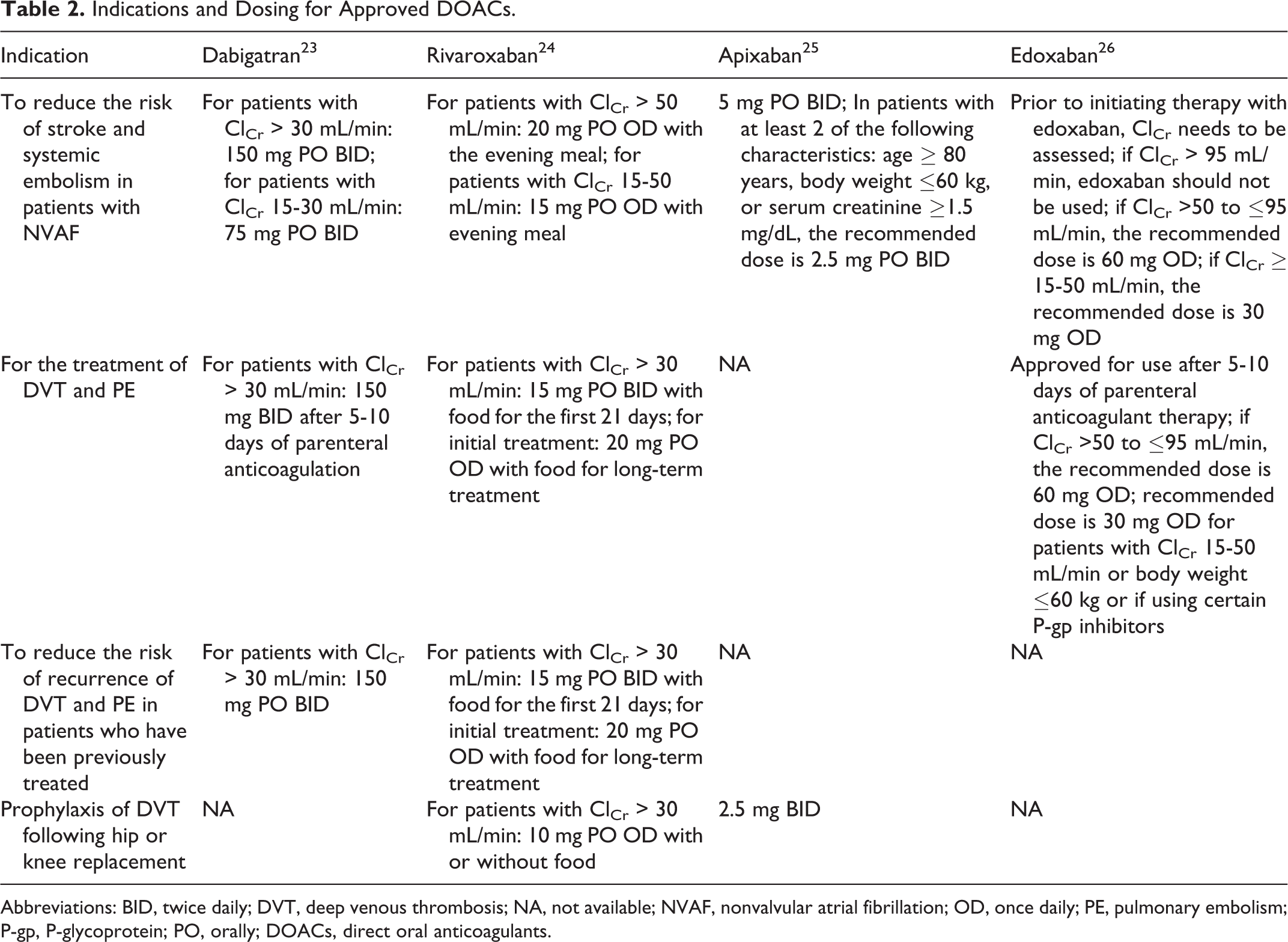
Abbreviations: BID, twice daily; DVT, deep venous thrombosis; NA, not available; NVAF, nonvalvular atrial fibrillation; OD, once daily; PE, pulmonary embolism; P-gp, P-glycoprotein; PO, orally; DOACs, direct oral anticoagulants.
Methodological Approach
This narrative review was developed on the basis of the authors’ clinical experience, their knowledge of relevant clinical and pharmacy literature, and their broad pharmacy experience managing patients with AF who are at risk for stroke and TE.
Individual Direct Oral Anticoagulants
A summary of the efficacy and safety results from pivotal clinical trials of the 4 approved direct oral anticoagulants (DOACs) can be found in Table 3.
Summary of Efficacy and Safety From Pivotal Clinical Trials for Approved DOACs.
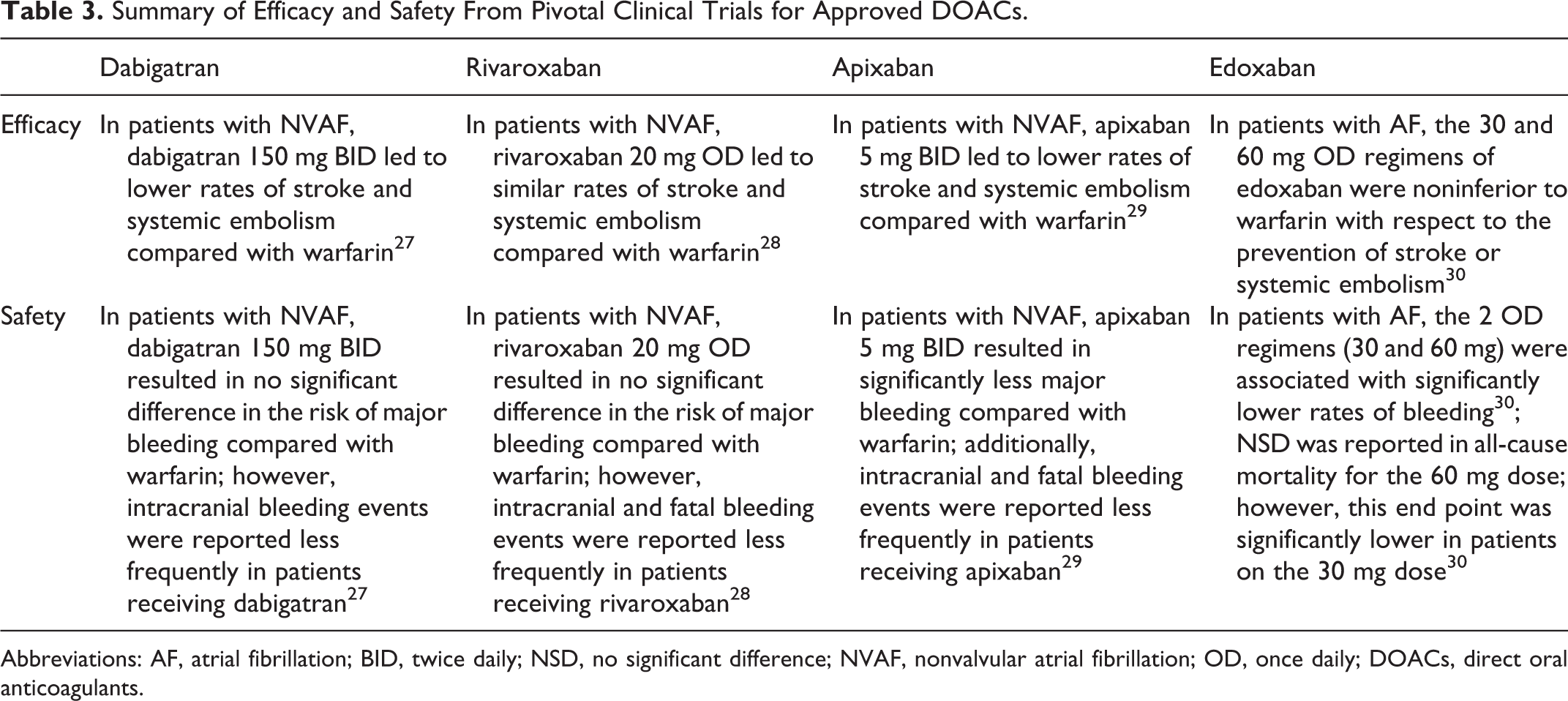
Abbreviations: AF, atrial fibrillation; BID, twice daily; NSD, no significant difference; NVAF, nonvalvular atrial fibrillation; OD, once daily; DOACs, direct oral anticoagulants.
Detailed below are drug and clinical profiles of these agents, dabigatran, rivaroxaban, apixaban, and the newly approved FXa inhibitor, edoxaban.
Dabigatran Etexilate (Pradaxa)
Dabigatran is a specific inhibitor of the last step of the coagulation pathway and was the first DOAC to be approved for clinical use by the US Food and Drug Administration (FDA). 23 The parent compound, dabigatran etexilate, is hydrolyzed into the active compound dabigatran by esterases in the plasma and liver. In the United States, the use of dabigatran is approved for the following indications: (1) in patients with NVAF to reduce the risk of stroke and systemic embolism; (2) for the treatment of deep venous thrombosis (DVT) and pulmonary embolism (PE) in patients receiving a parenteral anticoagulant for 5 to 10 days; (3) in patients who have been previously treated with anticoagulant therapy to reduce the risk of recurrence of DVT and PE; and (4) for the primary prophylaxis of DVT and PE in patients who have undergone hip replacement surgery. 23
Mechanism of Action
Dabigatran and its active acyl glucuronide metabolites are competitive, direct inhibitors of thrombin, the serine protease responsible for the conversion of fibrinogen into fibrin as the last step of the coagulation cascade (Figure 1). 23 Competitive inhibition of both the free and clot-bound forms of thrombin prevents the development and growth of a thrombus. Dabigatran and its active metabolites can also inhibit thrombin-induced platelet aggregation. 23 Consideration of key PK and PD parameters can help guide clinical decisions regarding the potential use of dabigatran in individual patients.
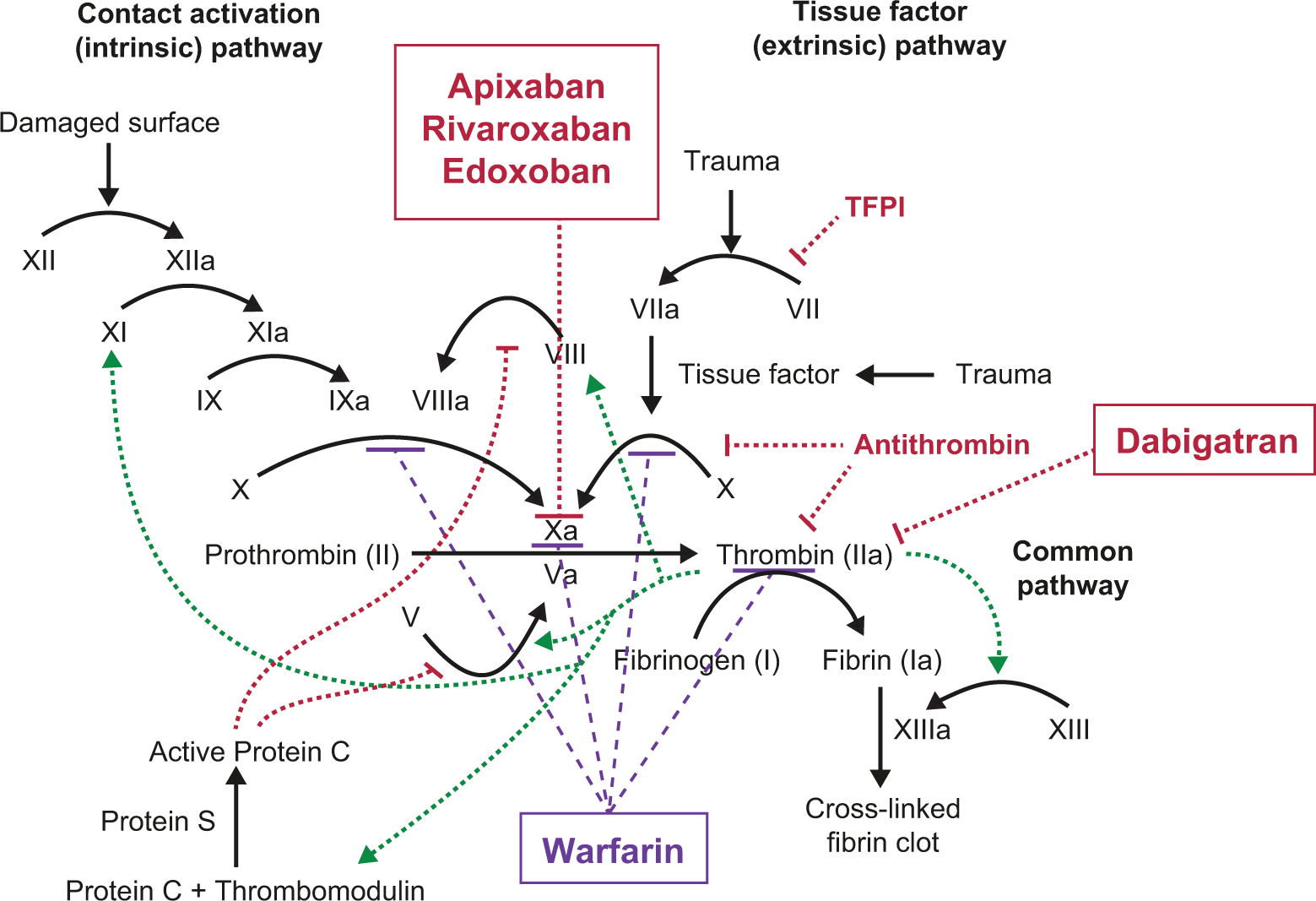
Clotting cascade with warfarin and approved direct oral anticoagulants (DOACs). The intrinsic, extrinsic, and common clotting pathways in the human body, and the sites of action of warfarin (whose inhibition of the synthesis of vitamin K-dependent proteins impacts clotting along several fronts), and those of the currently approved DOACs (rivaroxaban, apixaban, and edoxaban each inhibit factor Xa directly). Adapted from “Coagulation full” by Joe D, Own work. Licensed under Creative Commons Attribution-Share Alike 3.0 via Wikimedia Commons—http://commons.wikimedia.org/wiki/File: Coagulation_full.svg#mediaviewer/File: Coagulation_full.svg. Permission has been granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, in this Wikimedia link.
The PD properties of dabigatran include prolongation of coagulation times in assays such as the activated partial thromboplastin time (aPTT), ecarin clotting time (ECT), and thrombin time (TT). 23 As a marker of coagulation efficacy, the INR is relatively insensitive to inhibition by dabigatran. The half-life of dabigatran in healthy participants is 12 to 17 hours. 23
The proportional bioavailability of dabigatran per oral dose is in the range of 3% to 7%, and the etexilate prodrug is picked up by the efflux transporter P-glycoprotein (P-gp). In the fasting state, Cmax is reached 1 hour after dosing, while concurrent dosing with a high-fat meal can delay the Cmax by approximately 2 hours. Dosing with or without food does not change the overall dabigatran bioavailability. 23 Patients should be aware that dabigatran capsules need to be intact at dosing; oral bioavailability can increase if the capsules are compromised. There is moderate binding of dabigatran to human plasma proteins, of the order of ∼35%. 23
Dabigatran has been reported to interact with a number of other drugs that interact with the P-gp transporter on the gut level, leading to potential alterations in PK and PD parameters. Known drug–drug interactions with dabigatran include P-gp inducers such as rifampin, which when dosed at 600 mg/d for 7 days reduced the area under the curve (AUC) and Cmax of dabigatran by 66% and 67%, respectively. 23 On the other hand, P-gp inhibitors including ketoconazole, dronedarone, amiodarone, verapamil, and quinidine (with the exception of ketoconazole, these drugs are commonly prescribed for patients with AF) may increase the total exposure of dabigatran, however, only the first 2 may generally lead to clinically relevant changes in plasma concentrations of dabigatran. Thus, prescribers should consider a dose reduction to 75 mg twice daily, if a patient’s creatinine clearance (Clcr) is in the range of 30 to 50 mL/min, and the patient is also taking ketoconazole and/or dronedarone. No significant effects by dabigatran were observed on the metabolism of other drugs that interact with the CYP3A4 and CYP2C9 pathways (atorvastatin, clarithromycin, diclofenac, clopidogrel, and digoxin). 23
Dabigatran is contraindicated in patients with a history of pathological bleeding, any prior hypersensitivity reactions to dabigatran, or a mechanical prosthetic heart valve. 23 The most common adverse reactions, with an incidence of >15%, are gastritis-like symptoms and bleeding. Dabigatran capsules contain dabigatran etexilate and tartaric acid to improve the bioavailability of the medication. Of note, there is no recommended option for nasogastric tube administration of dabigatran.
A key benefit when considering dabigatran therapy is that for most patients, the extent of anticoagulation does not need to be assessed regularly. 23 If an estimate is needed, the aPTT or ECT, but not INR, should be used to monitor anticoagulant activity in patients, since the INR value does not correlate well with the level of anticoagulation provided by dabigatran. 23 The aPTT assay, if used with a full complement of centrally validated reagents, may be useful for semiquantitative assessments of dabigatran levels; since the assay is curvilinear for dabigatran concentrations, precise quantitation can be difficult to achieve in practice. 31 Currently, in the European Union, the diluted TT assay is recommended for quantitative determination of plasma levels of dabigatran. 32 Also, existing laboratory platforms may find novel applications for the quantitative assessments of anti-IIa and Xa anticoagulants, potentially allowing rapid and reliable quantitation of DOAC activity at a local level. 33 As these technologies become more widely used, they may provide additional information in cases where rapid assessment of DOAC-induced anticoagulation effects may be helpful.
Geriatric patients comprise a major category of patients who are candidates for dabigatran therapy, and in the pivotal RE-LY (Randomized evaluation of long term anticoagulant therapy) trial, 82% of the patients were ≥65 years of age, whereas 40% were ≥75 years. Stroke and bleeding risks are known to increase with older age, however, the risk–benefit profile of dabigatran is favorable in all geriatric age groups. 23
A number of pivotal clinical trials have been reported, and their results provide important insights into the potential clinical benefit of dabigatran. In the RE-LY trial, 18 113 patients with AF who were at risk for stroke (mean CHADS-2 score = 2.1) received dabigatran (110 or 150 mg twice daily, blinded) or warfarin (adjusted dose, unblinded). The primary outcome was stroke or systemic embolism. 27 After a median follow-up of 2 years, rates of the primary outcome were lower in both the dabigatran-treated groups: 1.54% per year for 110 mg dabigatran (RR: 0.90; 95% CI: 0.74-1.10; P = .29) and 1.11% per year for 150 mg dabigatran (RR: 0.65; 95% CI: 0.52-0.81; P < .001) versus 1.69% per year in the warfarin group. 34 The annual rates of major bleeding events were 3.61% for the warfarin group, 2.92% for the dabigatran 110 mg group (P = .003), and 3.40% for the dabigatran 150 mg group (P = .41). 34 As noted in an earlier analysis of the RE-LY trial data, the annual rates of intracranial bleeding were 0.74% for the warfarin group, 0.23% for the dabigatran 110 mg group (P < .001), and 0.30% for the dabigatran 150 mg group (P < .001). 27 In patients with AF, dabigatran therapy (150 mg) led to lower rates of stroke and systemic embolism, with similar rates of major hemorrhage (vs warfarin). 27
Due to potential underreporting of events, data from the RE-LY trial were reanalyzed in 2010 using multiple algorithms to identify any symptom of a primary or secondary event or bleeding. 35 As a result, 81 new events were identified in 80 patients, including 1 stroke, 1 systemic embolic event, 4 clinical myocardial infarctions (MIs), 1 PE, 5 TIAs, and 69 major hemorrhages. 35 Also, a review of the electrocardiogram reports revealed 28 cases whose reports were consistent with silent MI. 35 The reanalysis that included the newly identified events did not significantly change the study results, and the authors determined that the original conclusions were unchanged. 35
In a further reanalysis of the RE-LY data, more than 1500 cases (mainly cases of death) were reevaluated to determine whether a concurrent not yet reported event may have occurred. 34 The authors identified 20 cases with previously unreported events and 2 cases with additional major bleeding episodes, in addition to the deaths already reported. Of these 22 cases, there were 2 occurrences of stroke (1 in the group receiving 150 mg of dabigatran twice daily and 1 in the group receiving warfarin) and 20 episodes of major bleeding (5 in the dabigatran 110 mg twice daily group, 10 in the dabigatran 150 mg twice-daily group, and 5 in the warfarin group). 34 Major bleeding episodes were considered to be life threatening in 4 patients receiving 110 mg dabigatran, in 4 patients receiving 150 mg dabigatran, and in 3 patients receiving warfarin. These episodes were considered to be fatal in 3 patients receiving 110 mg dabigatran, in 2 patients receiving 150 mg dabigatran, and in 3 patients receiving warfarin. 34 All but 2 of these additional events were discovered in patients whose deaths were already reported. Based on the results of this new analysis, including the newly identified events, the authors determined that the original conclusions were unchanged. 34
In a very recent analysis of the relative safety of dabigatran for the treatment of NVAF in elderly patients seen in general practice settings, the propensity score-matched new user cohorts were identified from Medicare patients initiating dabigatran or warfarin for NVAF between October 2010 and December 2012. 36 From a total of 431 414 patients, a group of 134 414 were selected for this analysis, and there were data representing 37 587 person-years of follow-up, including 2715 primary outcome events (ischemic stroke, major bleeding with specific focus on intracranial and gastrointestinal (GI) bleeding, and acute MI). 36 The analysis determined the hazard ratios (HRs) plus 95% CIs for the following outcomes: ischemic stroke (0.80; 0.67-0.96), intracranial hemorrhage (0.34; 0.26-0.46), major GI bleeding (1.28; 1.14-1.44), acute MI (0.92; 0.78-1.08), and death (0.86; 0.77-0.96), when comparing treatment with dabigatran versus warfarin. 36
A related analysis of patients with NVAF from the US Department of Defense claims database (October 2009 to July 2013) looked at outcomes with either dabigatran or warfarin treatment. In this propensity score-matched cohort (n = 12 793 per group), adjusted HRs indicate that patients on dabigatran experienced fewer strokes (0.73; 95% CI: 0.55-0.97), major intracranial (0.49; 0.30-0.79) and other (0.38; 0.22-0.66) bleeding, MI (0.65; 0.45-0.95), and deaths (0.64; 0.55-0.74) than patients receiving warfarin. 37
Major bleeding events (0.87; 0.74-1.03) and major GI bleeding events (1.13; 0.94-1.37) were similar between groups, whereas major lower GI bleeding events were more frequent (1.30; 1.04-1.62) in patients receiving dabigatran. 37
As further “real-world” follow-up, the safety and efficacy of dabigatran versus warfarin for the prevention of stroke examined data from 2 commercial health insurance databases using a propensity score-matched sequential cohort design. Among 19 189 new users of dabigatran or warfarin, the pooled HR for stroke was 0.77 (95% CI: 0.54-1.09) and for major hemorrhages was 0.75 (0.65-0.87). 38 Also, this analysis identified that patients on dabigatran experienced a lower rate of major bleeding events among patients aged <55 (HR = 0.51; 0.30-0.87) and with CHADS2 < 2 (HR = 0.58; 0.44-0.77). 38
The results of these observational studies are consistent with the data reported from the RE-LY trial, and they confirm the benefits of dabigatran versus warfarin in real-world settings. The prescription costs of dabigatran are estimated to be ∼ US$320/month or ∼US$3840/year per patient. 26
Rivaroxaban (Xarelto)
Rivaroxaban is a direct inhibitor of FXa, ultimately leading to a reduction in the downstream levels of activated thrombin that are available for thrombus formation (Figure 1). 39 Rivaroxaban has been approved by the US FDA for the following indications: (1) for reducing the risk of stroke and systemic embolism in patients with NVAF; (2) for the treatment of DVT and PE and for the reduction in the risk of recurrence of DVT and of PE; and (3) for the prophylaxis of DVT (possibly leading to PE) in patients undergoing knee or hip replacement surgery. 24 As a selective inhibitor of FXa, therapy with rivaroxaban leads to the inhibition of free FXa and the activation of prothrombinase. Rivaroxaban can also indirectly inhibit the platelet aggregation that is induced by thrombin (since FXa inhibition reduces levels of active thrombin). 24
The elimination half-life of rivaroxaban is 5 to 9 hours in healthy participants and 11 to 13 hours in the elderly patients. The bioavailability of rivaroxaban is dose dependent; for the 20 mg dose, it is approximately 66%. However, at this dosage, the mean AUC and Cmax increase by 39% and 76%, respectively, when taken with food. 24 Both the 15 and 20 mg doses should be administered with food. The PD parameters of rivaroxaban, including the selective inhibition of FXa activity, and the prolongation of prothrombin time (PT) as a key measure of the anticoagulant effects of rivaroxaban are progressive in a predictable and dose-dependent manner. 24
Dosing recommendations for rivaroxaban depend on the therapeutic goals and the patient. 24 For patients with NVAF and a Clcr > 50 mL/min, rivaroxaban should be dosed at 20 mg once daily with an evening meal. For patients with renal impairment and a Clcr of 15 to 50 mL/min, the dose should be 15 mg once daily with an evening meal. 24 For the treatment of DVT and PE, for the reduction in recurrent DVT, and for patients at risk for PE, the initial treatment regimens should align with the following recommendations: for initial treatment of acute DVT or PE, the dose should be 15 mg twice daily with food for the first 21 days; 24 for continued treatment, and to achieve long-term reductions in recurrence risk of DVT and of PE, the dose should be 20 mg once daily with food 24 ; for the prophylaxis of DVT after hip or knee replacement surgery, the dose should be 10 mg once daily with or without food. 24
Rivaroxaban has not demonstrated the induction of CYP1A2, 2B6, 2C19, or 3A4 as measured by in vitro assays. 24 Concomitant use of rivaroxaban with combined CYP3A4 and P-gp inhibitors (such as ketoconazole, itraconazole, lopinavir/ritonavir, ritonavir, indinavir, and conivaptan) 24,40 can increase rivaroxaban exposure, leading to proportional increases in PD effects (ie, FXa inhibition and PT prolongation) and potentially leading to an increase in bleeding risk. However, no formal recommendations for dose modification are provided by the manufacturer.
Contraindications for rivaroxaban include active pathologic bleeding and any history of a severe hypersensitivity reaction to rivaroxaban. The most common (>5%) adverse reaction reported with rivaroxaban was bleeding. After discontinuation of rivaroxaban, there is an increased risk of stroke in patients with NVAF, 24 as is true for patients who discontinue use of any of the currently approved novel oral anticoagulants.
Rivaroxaban can be administered via nasogastric tube when necessary. In an open-label crossover study, healthy volunteers received 20 mg of rivaroxaban as a whole tablet administered orally (reference dose), as a crushed tablet in applesauce (crushed-oral), or crushed tablet in water via nasogastric tube (crushed-NG). A standard 100 mL liquid meal followed each dosing event. 41 Standard Cmax and AUC parameters were estimated based on measured plasma concentrations of rivaroxaban. 41 When comparing crushed-oral to reference, the geometric mean ratios for Cmax and AUC∞ were 90.0% and 95.4%, respectively, and comparing crushed-NG to reference, these ratios were 82.0% and 89.1%, respectively, indicating that all 3 administration methods achieved bioequivalent plasma concentrations. 41
Pivotal clinical trials have established the efficacy of rivaroxaban. In the ROCKET-AF (Rivaroxaban once-daily oral direct factor Xa inhibition compared with vitamin K antagonism for prevention of stroke and embolism trial in atrial fibrillation) trial, 14 264 patients with NVAF at increased risk for stroke (mean CHADS2 score = 3.5) received either rivaroxaban (20 mg once daily) or dose-adjusted warfarin in a double-blind clinical study. The primary end point was stroke or systemic embolism, which by per-protocol, as-treated primary analysis, was reported in 188 patients (1.7% per year) receiving rivaroxaban and in 241 patients (2.2% per year) receiving warfarin (HR for rivaroxaban: 0.79; 95% CI: 0.66-0.96; P < .001 for noninferiority). 28 By intent-to-treat analysis, the primary end point occurred in 269 patients (2.1% per year) receiving rivaroxaban and in 306 patients (2.4% per year) receiving warfarin (HR: 0.88; 95% CI: 0.74-1.03; P < .001 for noninferiority and P = .12 for superiority). 28 Clinically relevant bleeding (major and nonmajor) was reported in 1475 patients (14.9% per year) receiving rivaroxaban and in 1449 (14.5% per year) receiving warfarin (HR: 1.03; 95% CI: 0.96-1.11; P = .44). Patients receiving rivaroxaban experienced significant reductions in intracranial hemorrhage (0.5% vs 0.7%; P = .02) and fatal bleeding (0.2% vs 0.5%; P = .003). 28 The prescription costs of rivaroxaban are estimated to be ∼US$310/month or ∼US$3720/year per patient. 26
Apixaban (Eliquis)
Apixaban is a selective FXa inhibitor that received US FDA approval for DVT prophylaxis following hip or knee replacement surgery and to reduce the risk of stroke and systemic embolism in patients with NVAF. 25 This drug is a reversible and selective inhibitor of the active site of the FXa. Apixaban has been shown to inhibit the activity of both free and clot-bound FXa; it also inhibits prothrombinase activity. Administration of apixaban also leads to the inhibition of platelet aggregation induced by thrombin, via indirect inhibition of thrombin formation. 25
Apixaban has predicable PK properties, with an absolute bioavailability of ∼50%; food intake does not appear to affect absorption. 25 The Cmax of apixaban in the blood is achieved within 3 to 4 hours after oral dosing. 25 Apixaban has a half-life of ∼12 hours after oral administration and demonstrates linear PK properties. 25 The PD characteristics of apixaban include prolongation of PT, INR, and aPTT via direct and specific inhibition of FXa. 25 Although changes in the results of these assays can be expected at therapeutic doses, they are generally small and highly variable, and therefore of limited utility for making clinical decisions regarding the anticoagulation effect of apixaban. 25 The majority of adult patients with NVAF should receive an oral dose of 5 mg twice daily. For patients who have 2 or more of the following characteristics, the recommended dosing is 2.5 mg twice daily (age ≥80 years, weight ≤60 kg, or serum creatinine level ≥1.5 mg/dL). 25
Drug–drug interactions are a potential concern, especially when supratherapeutic effects of an agent can lead to serious bleeding. However, as is true for all of the DOACs, the number of clinically significant drug interactions with apixaban is limited. Apixaban is not a significant inhibitor of P-gp. 25 However, apixaban is a substrate of P-gp and CYP3A4 and should be dose reduced (2.5 mg twice daily) when used with strong inhibitors of these enzyme systems such as clarithromycin and ketoconazole. In addition, apixaban should be avoided with concomitant strong P-gp and CYP3A4 inducers such as rifampin and phenytoin. 25
Apixaban is contraindicated in patients with active pathological bleeding or a history of severe hypersensitivity to the agent. 25 The adverse reactions of greatest concern reported with apixaban are serious to fatal bleeding, hypersensitivity reactions that include skin rashes, anaphylactic reactions (allergic edema), and syncope, all reported at rates of <1%. 25 The most common adverse reactions are gingival bleeding, epistaxis, contusion, hematuria, rectal hemorrhage, hematoma, menorrhagia, and hemoptysis, which have all been reported at >1%. 25 At present, nasogastric tube administration is not a recommended route of administration for apixaban.
The randomized, double-blind ARISTOTLE (Apixaban for reduction in stroke and other thromboembolic events in atrial fibrillation) trial compared apixaban (5 mg twice daily) with warfarin (target INR 2.0-3.0) in 18 201 patients with AF and at least 1 additional stroke risk factor (mean CHADS2 score = 2.1). 42 The primary outcome of the trial was ischemic or hemorrhagic stroke or systemic embolism. With a median follow-up of 1.8 years, primary outcome rates were 1.27% versus 1.60% per year (apixaban vs warfarin, respectively) with an HR for apixaban of 0.79 (95% CI: 0.66-0.95; P < .001 for noninferiority and P = .01 for superiority). 42 Rates of major bleeding events were 2.13% versus 3.09% per year (apixaban vs warfarin, respectively), with an HR for apixaban of 0.69 (95% CI: 0.60-0.80; P < .001). Deaths from any cause occurred at rates of 3.52% and 3.94%, respectively (HR: 0.89; 95% CI: 0.80-0.99; P = .047). 42 Reported rates of intracranial hemorrhage were 0.33% versus 0.80% per year (apixaban vs warfarin, respectively), with an HR for apixaban of 0.42 (95% CI: 0.30-0.58; P < .001). 42
Results from several major clinical trials have provided evidence that apixaban may be a reliable alternative for patients who are not suitable candidates for (or are unwilling to receive) VKA therapy. The prescription costs of apixaban are estimated to be ∼US$320/month, or ∼US$3840/year per patient. 26
Edoxaban (Savaysa)
Edoxaban is a selective inhibitor of FXa, and this inhibition leads to a reduction in thrombin generation and thrombus formation. Edoxaban also does not depend on antithrombin III to exert this antithrombotic effect. Edoxaban inhibits free FXa, prothrombinase activity, and thrombin-induced platelet aggregation. 43
Edoxaban has a terminal elimination half-life of 10 to 14 hours following oral administration and demonstrates PKs that are approximately proportional to the dose, in the range of 15 to 150 mg. 43 Edoxaban is primarily eliminated via the kidneys as unmodified drug, which accounts for approximately 50% of total drug clearance. The remaining fraction is eliminated via a combination of drug metabolism and biliary/intestinal excretion. 43
Edoxaban has reduced efficacy in patients with NVAF having Clcr >95 mL/min and therefore should not be used in this patient population. 43 Edoxaban does prolong the PT and aPTT. However, in the therapeutic dose range, the changes observed in the PT, INR, and aPTT are minor, subject to considerable variability, and ultimately not clinically useful. 43
The ENGAGE AF-TIMI 48 (Effective anticoagulation with factor Xa next generation in atrial fibrillation thrombolysis in myocardial infarction 48) was a randomized, double-blind trial to examine the long-term efficacy and safety of once-daily regimens of edoxaban (low dose 30 mg or high dose 60 mg), or warfarin (dose adjusted to an INR of 2-3), in 21 105 patients with AF. 30 The primary efficacy end point was stroke or systemic embolism, and the principal safety end point was major bleeding. 30
The primary end point was reported at a rate of 1.50% per year with warfarin, compared with 1.18% for high-dose edoxaban (HR: 0.79; 97.5% CI: 0.63-0.99; P < .001 for noninferiority) and 1.61% with low-dose edoxaban (HR 1.07; 97.5% CI: 0.87-1.31; P = .005 for noninferiority). 30 Rates of major bleeding were 3.43% per year with warfarin versus 2.75% per year with high-dose edoxaban (HR: 0.80; 95% CI: 0.71-0.91; P < .001) and 1.61% per year with low-dose edoxaban (HR: 0.47; 95% CI: 0.41-0.55; P < .001). 30
All-cause mortality rates were 4.35% per year with warfarin versus 3.99% per year with high-dose edoxaban (HR: 0.92; 95% CI: 0.83-1.01; P = .08) and 3.80% per year with low-dose edoxaban (HR: 0.87; 95% CI: 0.79-0.96; P = .006). 30 For the prevention of stroke or systemic embolism, the results of this trial indicate that the 2 doses of once-daily edoxaban were noninferior to once-daily warfarin. 30 The prescription costs of edoxaban are estimated to be ∼US$300/month or ∼US$3600/year per patient. 26
Clinical Considerations and Gaps
How Should DOACS Be Used in Patients With Kidney Disease/Hepatic Impairment?
As a consequence of the major pathways of elimination of DOACs including both the renal and hepatic routes, special considerations should guide the use of DOAC agents in patients with kidney disease and/or hepatic impairment.
Dabigatran is a viable option for the subpopulation of patients who have moderate renal impairment (ClCr 30-50 mL/min). In these patients, the dabigatran half-life (T1/2) will increase to 18 hours, with a 3.2-fold increase in AUC and 1.7-fold increase in Cmax. 23 Larger changes in PK parameters (T1/2 = 27 hours, AUC = 6.3-fold, and Cmax = 2.1-fold) are predicted based on PK modeling for patients with severe renal disease (ClCr 15-30 mL/min), who consecutively should receive a lower dabigatran dose of 75 mg twice daily. 23 For patients with moderate liver disease (Child-Pugh B), dabigatran PK results demonstrated considerable intersubject variability, however, no definite change in exposure or PD was noted. 23
Similarly, following a single 10 mg dose, rivaroxaban exposure (AUC) was elevated in participants with renal impairment (up to ∼26% in patients with ClCr of 15-29 mL/min) versus healthy participants with a normal Clcr, and FXa inhibition was elevated by 12% in these same participants. 24 Rivaroxaban exposure (AUC) also was significantly elevated by 15% to 127% in patients with moderate hepatic impairment (Child-Pugh B) compared with healthy participants having normal liver function. These patients also had a 27% increase in Cmax. 24
Apixaban can be considered for patients with either moderate renal disease or hepatic impairment, since AUC and Cmax parameters for apixaban in these patient populations were within 1.5-fold of reference values. 25 For patients on hemodialysis due to end-stage renal disease, the recommended dose is 5 mg twice daily based on PK and PD studies using anti-FXa activity levels. 25 This dose should be reduced to 2.5 mg twice daily if the patient is either ≥80 years of age or if the body weight is ≤60 kg. 25 For patients with mild hepatic impairment, no dose adjustment is needed. However, it is important to note that apixaban is not recommended for patients with severe hepatic impairment. 25
Protocols for Interrupting DOAC Regimens in Advance of Medical/Surgical Procedures
The appropriate management of patients during the perioperative period is another important clinical challenge that warrants special monitoring and dosing considerations. To better mitigate potential bleeding events, when invasive procedures or surgery are being considered, dabigatran therapy should be discontinued 1 to 2 days prior to the procedure (for patients with ClCr ≥ 50 mL/min) or 3 to 5 days in advance (for patients with ClCr < 50 mL/min). 23 However, for patients undergoing major surgical procedures or spinal puncture or for those who will receive a catheter (spinal or epidural) or port, where complete hemostasis may be necessary, longer wait times should be considered. 23
For patients receiving rivaroxaban, therapy should be terminated at least 24 hours in advance of the procedure. 24 In addition, the RRs associated with any potential increase in bleeding events should be weighed against the urgency of the medical/surgical intervention. Once the patient is stable, then rivaroxaban should be reinitiated as soon as adequate hemostasis has been established following the immediate procedure. An epidural catheter should not be removed less than 18 hours from the previous rivaroxaban dose, and the next rivaroxaban dose should not be administered within 6 hours following catheter removal.
Apixaban should be discontinued at least 48 hours in advance of elective surgery or invasive procedures that are associated with moderate or high risk of significant bleeding. 25 Discontinue apixaban at least 24 hours in advance of elective surgery or invasive procedures associated with low risk of bleeding, or where the bleeding would be easily controlled, and in a noncritical location. 25 As soon as appropriate hemostasis has been established after the procedure, apixaban should be reinitiated. An epidural catheter should not be removed less than 24 hours from the previous apixaban dose, and the next rivaroxaban dose should not be administered within 5 hours following catheter removal.
Protocols to Better Manage Bleeding Events in DOAC-Treated Patients
A key limitation of the DOACs has been the lack of approved, specific reversal agents for use when emergency surgeries or life-threatening bleeding events arise. This unmet medical need has driven the ongoing investigation and the development of specific reversal agents and also nonspecific protocols that rely on supportive care and general clotting factors to reverse the anticoagulation effects of individual DOACs.
In a randomized, double-blind, placebo-controlled study, 6 healthy male volunteers received rivaroxaban 20 mg twice daily for 2.5 days, followed by either a single bolus of 50 IU/kg prothrombin complex concentrate (PCC, Cofact; Sanquin Blood Supply, Amsterdam, the Netherlands) or similar volume of saline. 44 Rivaroxaban induced a significant prolongation of the PT (15.8 ± 1.3 seconds vs 12.3 ± 0.7 seconds at baseline; P < .001). This effect was immediately and completely reversed by PCC (12.8 ± 1.0 seconds; P < .001). 44 The anticoagulant efficacy of rivaroxaban is not reliably measured with standard laboratory tests, and as is currently the case for all DOACs, it cannot be easily reversed. Therefore, health-care providers should promptly evaluate the significance of clinical signs or symptoms that are consistent with potential blood loss (lower hemoglobin and/or hematocrit red, hypotension, or fetal distress). 24
No specific coagulation tests are recommended to accurately assess the degree of the anticoagulation achieved with apixaban therapy. In addition, no specific agent has been approved for clinical use to reverse the anticoagulant effect of apixaban, to date. 25 However, a specific reversal agent is currently under development (described subsequently). Per the product label, activated charcoal, administered soon after apixaban dosing (2-6 hours), may assist in the management of apixaban overdose or accidental ingestion. 25
Specific reversal agents for individual DOAC types are currently under clinical development and review by the FDA. To reverse the anticlotting effect of dabigatran, a specific monoclonal antibody (idarucizumab) that binds dabigatran with high affinity, and therefore relieves the inhibition of thrombin, has been the focus of a number of clinical trials where it has demonstrated a rapid and sustained reversal of prolongations in all clotting assays studied (TT, aPTT, ECT, and activated clotting time). 45 After breakthrough therapy designation from the US FDA, 46 where it was assigned to an accelerated track for review, idarucizumab was recently approved by the US FDA for clinical use to reverse the anticoagulation effect of dabigatran for emergency surgeries or procedures or in case of life-threatening or uncontrolled bleeding. 47 In addition, in cases where emergency surgery and/or urgent procedures may be required, or when a patient has life-threatening or uncontrolled bleeding, idarucizumab has just been approved by the European Commission to rapidly and specifically reverse the anticoagulant effects of dabigatran. 48
For FXa inhibitors, such as rivaroxaban and apixaban, a recombinant human FXa molecule, andexanet alfa, has been developed, which retains the high-affinity motif that binds the FXa inhibitors, but andexanet alfa lacks the catalytic activity of native FXa, and therefore acts as a decoy for the FXa inhibitor anticoagulants, leading to reversal of their effects. 49
Protocols for Switching a Patient Between Available DOACs
A common clinical question relates to the recommendations for conversion between DOAC and warfarin therapies. Protocols for converting a patient to or from warfarin and individual DOAC agents have been developed based on clinical experience, and these are specific to the particular conversion in question. Recommendations for individual conversion protocols are presented and summarized in Table 4.
Recommendations for Conversion Between Various Anticoagulants.
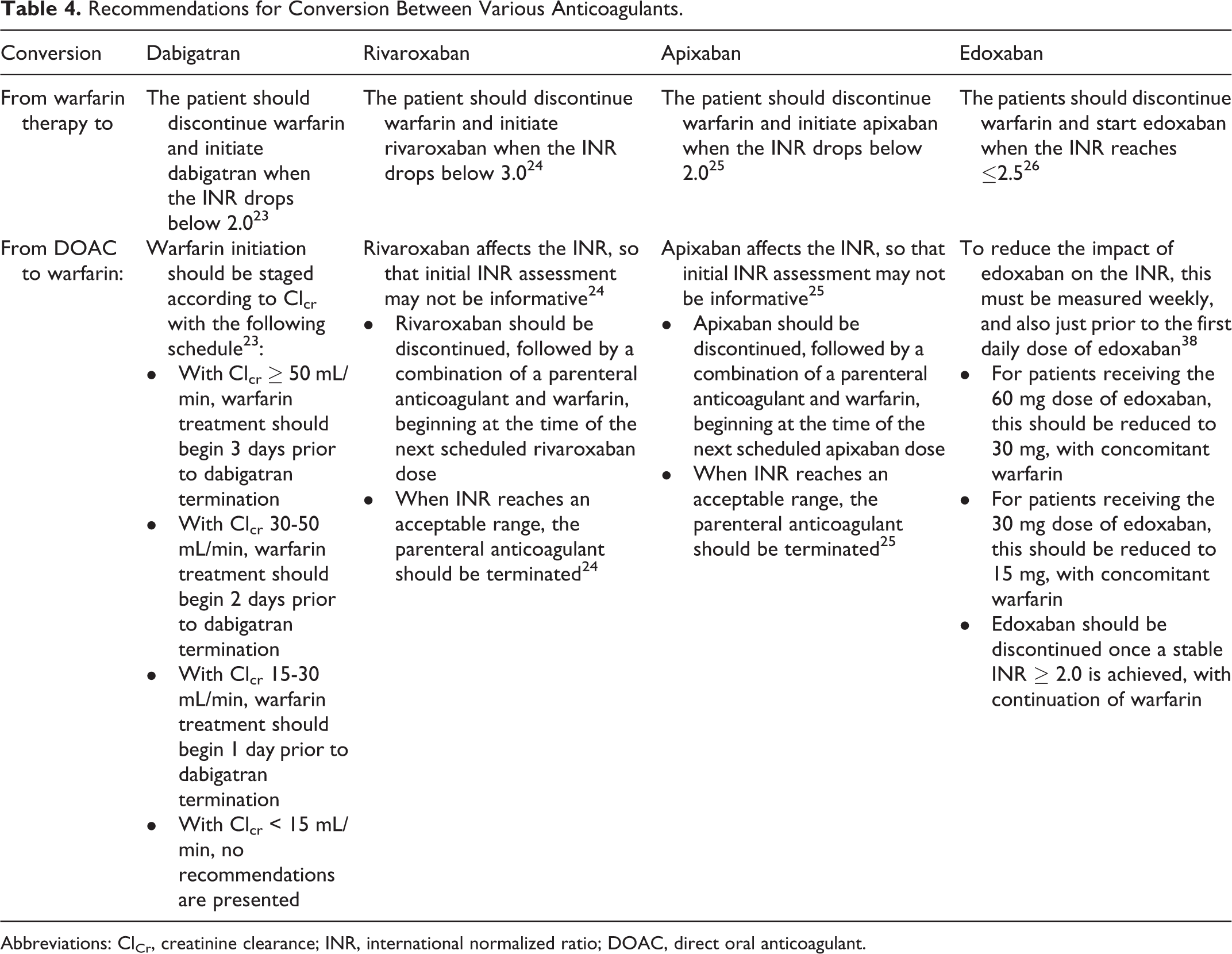
Abbreviations: ClCr, creatinine clearance; INR, international normalized ratio; DOAC, direct oral anticoagulant.
Clinical Risks Associated With DOAC Therapy
Concerns have been raised about other potential nonbleeding risks in patients taking DOACs. For instance, potential increases in the risk for MI have been studied in dabigatran-treated patients. In the RE-LY trial, the annual rates of MI were higher with dabigatran (0.82%/year with 110 mg twice daily; 0.81%/year with 150 mg twice daily) compared with warfarin (0.64%/year), with an HR of 1.29 (95% CI: 0.96-1.75; P = .09) for dabigatran 110 mg and an HR of 1.27 (95% CI: 0.94-1.71; P = .12) for dabigatran 150 mg. 50
In a recent meta-analysis of results from 14 randomized clinical trials compared with warfarin-treated controls, the Peto odds ratio for the dabigatran-treated patients based on a fixed-effect model for MI was 1.41 (95% CI: 1.11-1.80; P = .005), for other cardiovascular events was 0.94 (95% CI: 0.83-1.06; P = .293), for major bleeding was 0.85 (95% CI: 0.76-0.96; P = .007), and for all-cause mortality was 0.90 (95% CI: 0.81-1.01; P = .061). 51 For the analysis of MI risk, data from 12 trials were available, which reported MI in 218 (1.31%) of 16 686 patients on dabigatran and 80 (0.79%) of 10 157 patients in the control arm. 51 This analysis demonstrated an increased risk for MI but lower risks for major bleeding events. Other parameters, including other cardiovascular events and all-cause mortality rates, were unchanged.
A separate analysis of data from 14 comparative trials (n = 42 484) looking at the occurrence of cardiovascular events in patients treated for 5 different indications included comparisons of dabigatran with warfarin, enoxaparin, and placebo. 52 Pooled analysis comparing dabigatran with warfarin for MI demonstrated a modest benefit for warfarin, with odds ratio (OR) of 1.30 (95% CI: 0.96-1.76 for dabigatran 110 mg twice daily) and 1.42 (95% CI: 1.07-1.88 for dabigatran 150 mg twice daily). 52 Analysis by the clinically relevant composite end point of MI, total stroke, and vascular death demonstrated numerically fewer events in patients receiving dabigatran (150 mg twice daily), with an OR of 0.87 (95% CI: 0.77-1.00), and was similar for patients receiving dabigatran (110 mg twice daily), with an OR of 0.99 (95% CI: 0.87-1.13). 52 The pooled analysis demonstrated that patients receiving dabigatran had similar MI rates to those receiving either enoxaparin or placebo.
On the other hand, a major study by the US FDA of 134 000 Medicare patients, aged 65 years and older, compared outcomes in patients with NVAF being treated with dabigatran versus warfarin. No difference in the rates of MI between the dabigatran-treated patients (for both 75 and 150 mg doses combined) and warfarin-treated patients was seen in this analysis (adjusted HR: 0.92; 95% CI: 0.78-1.08) for dabigatran versus warfarin. 53
Similarly, although rivaroxaban has been demonstrated to be noninferior to warfarin for the prevention of stroke in patients with AF, emergent concerns have been raised with regard to possible increases in stroke risk soon after rivaroxaban discontinuation. In a post hoc analysis of data from the ROCKET-AF trial, results from 14 624 patients were analyzed for stroke or non-central nervous system (CNS) embolism within 30 days after temporary interruptions of 3 days or more, early permanent study drug discontinuation, and end-of-study transition to open-label therapy. After temporary interruptions in therapy, stroke, and non-CNS embolism, events were reported at similar rates (rivaroxaban: n = 9, warfarin: n = 8; 6.20 vs 5.05/100 patient-years; HR: 1.28; 95% CI: 0.49-3.31; P = .62) and after early permanent discontinuation (rivaroxaban: n = 42, warfarin: n = 36; 25.60 vs 23.28/100 patient-years; HR: 1.10; 95% CI: 0.71-1.72; P = .66). When switching to open-label therapy after the study, patients who had been receiving rivaroxaban had more strokes (n = 22) than patients who had received warfarin (n = 6; 6.42 vs 1.73/100 patient-years; HR: 3.72; 95% CI: 1.51-9.16; P = .0044). No significant difference was noted between the treatment groups in all thrombotic events occurring within 30 days after termination of any study drug. The increased risk of stroke and non-CNS embolism observed in rivaroxaban-treated patients after permanent discontinuation of study drug highlights the need to ensure continued anticoagulation during any therapeutic transition period.
At the end of the ARISTOTLE study, an increased risk of stroke was observed when patients on apixaban were switched to alternative agents, usually a VKA. To effect this transition at the end of the study, patients generally received both apixaban and a VKA for 2 days. Over the subsequent 30 days following the end of the study, in the 6791 patients on apixaban, there were 21 stroke or systemic embolism events (0.3%), compared with 5 (0.1%) of the 6569 patients on warfarin. One interpretation of this result is that the patients may not have been on warfarin for enough time to attain a stable, therapeutic INR before terminating apixaban therapy. 25
Adherence Issues in Patients Who Are Receiving DOAC Therapies
Treatment adherence is a concern with any drug regimen that requires regular and chronic dosing, with patient motivation and treatment tolerability playing important roles in patient adherence to a treatment program over time. With regard to patients who may not be regularly adherent to their medications, anticoagulant therapy with warfarin may be the better choice owing to the longer half-life of the associated anticoagulant effects. On the other hand, patients who are nonadherent to the necessary regular monitoring of warfarin anticoagulation may be better candidates for DOAC therapy because regular monitoring is not necessary.
A recent meta-analysis of data from 18 randomized clinical trials, which included a total of 101 801 patients, reported that there was no statistically significant difference in the discontinuation rates between DOACs and pharmacologically active comparators in patients being treated for venous thromboembolism/PE risk (RR: 0.91; 95% CI: 0.74-1.13; P = .40). 54 There was also no significant difference between the discontinuation rates for DOACs compared with warfarin and aspirin for the prevention of stroke in patients with AF (RR: 1.01; 95%: CI 0.87-1.17; P = .92). 54 However, in patients with acute coronary syndromes, discontinuations of study drugs were significantly higher with DOACs than with placebo (RR: 1.40; 95% CI: 1.07-1.83; P = .01). 54 It should be noted that the overall discontinuation rates of DOACs were comparable in this patient population with the rates seen in patients receiving active comparators.
Conclusion
For many years, warfarin has been the mainstay oral option for stroke prophylaxis in patients with AF. Over the past 5 years, however, the DOACs have emerged as alternative options for patients with AF as well as other conditions requiring anticoagulation. The predictable PKs and relatively favorable drug and food interactions associated with these newer agents offer several benefits over traditional anticoagulation therapy with warfarin. However, limitations with these newer agents remain in regard to reversal and monitoring. As the body of literature continues to grow with the use of DOACs, there is hope for optimized treatment regimens and improved patient care.
Footnotes
Authors’ Note
The authors meet all criteria for authorship as recommended by the International Committee of Medical Journal Editors, were fully responsible for all content and editorial decisions, and were involved at all stages of manuscript development.
Acknowledgments
The authors acknowledge the writing and editorial assistance of José L. Walewski, PhD, of Envision Scientific Solutions, whose services were funded by Boehringer Ingelheim Pharmaceuticals, Inc. (see end of manuscript for full acknowledgments).
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Dr. Stacy has provided consulting services for Janssen Pharmaceuticals.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Editorial services were funded by Boehringer Ingelheim Pharmaceuticals, Inc.