
Abstract
Hematopoietic stem cell transplant-associated thrombotic microangiopathy (TA-TMA) is a fatal, multifactorial disorder, which may present with thrombocytopenia, hemolysis, acute renal failure, mental status changes and involvement of other organs. The pathogenesis of TA-TMA is complex and includes multiple risk factors such as certain conditioning regimens, calcineurin inhibitors (CNIs), graft-versus-host disease (GVHD), human leukocyte antigen mismatch, and opportunistic infections. The end result of these insults is endothelial injury in the kidney and other organs. Recent studies also indicate a role of complement activation in tissue damage. The lack of sensitive and specific diagnostic tests for TA-TMA often results in delayed diagnosis. Biopsy is not always possible for diagnosis because of the risk of complications such as bleeding. Recently, an emerging role of renal-centered screening approach has been demonstrated, which utilize the monitoring of blood pressure, urine protein, serum lactate dehydrogenase and hemogram for early detection. Therapeutic options are limited, and plasma exchange plays a minor role. Withdrawal of offending agent such as CNIs and the use of rituximab can be effective in some patients. However, the current treatment strategy is suboptimal and associated with high mortality rate. Recently, eculizumab has been utilized in a few patients with good outcomes. Patients, who develop TA-TMA, are also at an increased risk of GVHD, infection, renal, cardiovascular, and other complications, which can contribute to high mortality. Better understanding of molecular pathogenesis, improvement in posttransplant management, leading to early diagnosis, and management of TA-TMA are required to improve outcomes of this fatal entity.
Keywords
Introduction
Hematopoietic stem cell transplant (HSCT)-associated thrombotic microangiopathy (TA-TMA) is a fatal, multifactorial disorder. Similar to other TMAs such as thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic uremic syndrome (aHUS), it manifests with thrombocytopenia, Coombs-negative hemolysis, schistocytes on peripheral blood, acute renal failure, mental status changes, and involvement of other organs. 1,2 Incidence of TA-TMA after transplant is variable and may be associated with a variety of transplant-related factors such as conditioning regimens, immunosuppressive agents, graft-versus-host disease (GVHD), human leukocyte antigen (HLA) mismatch, and opportunistic infections. 3 –6 Some investigators have raised the possibility that TA-TMA may be considered as the renal involvement in acute and chronic GVHD. 5,7,8 The TA-TMA, which has a higher incidence in allogenic HSCT than autologous HSCT, occurs in as many as 10% to 20% of patients with allogeneic HSCT. It develops as early as 1 week to as late as after 1 year following HSCT; however, most of the patients are diagnosed within the first 3 months. 2 –6
Risk Factors and Pathogenesis
The major risk factors for TA-TMA include acute GVHD, use of conditioning with total body irradiation, advanced age, female sex, advanced primary hematologic malignancy, use of unrelated donor transplant, nonmyeloablative transplant, high-dose busulfan, and HLA mismatch. 9
The pathogenesis of TA-TMA is complex and involves multiple causative factors, which ultimately lead to endothelial injury in kidney and other organs, the final “common pathway” in TA-TMA. 10 Cohen et al demonstrated absent endothelial prostacyclin release and features of endothelial damage on electron microscopy. 11 In addition, von Willebrand factor (vWF) antigen levels are elevated with normal vWF multimer patterns, signifying endothelial injury. Elevated levels of interleukin (IL) 1, tumor necrosis factor α (TNF-α), interferon γ (IFN-γ), and IL-8, which may directly cause endothelial toxic damage or enhance inflammation-mediated tissue injury, have been reported. 12 Elevated levels of other plasma markers associated with endothelial injury, namely, thrombomodulin (TM), plasminogen activator inhibitor 1(PAI-1), and soluble intercellular adhesion molecule 1(ICAM-1) have been reported. 10
The primary etiology in TA-TMA, in contrast to TTP, is not deficiency of ADAMTS13. 9 Patients with TA-TMA usually have >10% ADAMTS13 serum activity, which may explain the lack of response to plasma exchange in the majority of these cases. Recent evidence demonstrates that complement pathways may become activated in cases with TA-TMA resulting in tissue damage. 13 Direct endothelial tissue damage from triggers such as high-dose chemotherapy, calcineurin inhibitors (CNIs), infection with different viruses, and GVHD can lead to classic complement activation. There have been reports of arteriolar C4d deposits in patients with TA-TMA after HSCT. 14 In alternative pathway, there is formation of the pathogenic autoantibody against complement factor H (CFH) due to either defects in CFHR3-CFHR1 genes associated with defective or absent CFHR1 protein or immune dysregulation after HSCT without genetic predisposition. 15
Immunosuppressive drugs play an important role in the pathogenesis of TA-TMA. The use of CNIs and sirolimus may directly damage endothelium or may be associated with activation of the alternative complement pathway. 9 Cyclosporine can also reduce levels of ADAMTS13 by inhibiting its secretion or by releasing vWF multimers that can form complexes with ADAMTS13. 16 Infections with several microorganisms such as Aspergillus, cytomegalovirus (CMV), and adenovirus have also been implicated in TA-TMA. 7,17 –20 Although no direct mechanism has been proposed, infections are a major activator of the alternate complement pathway, which may cause TA-TMA. 9 Similarly, there is a close association between TA-TMA and GVHD. Higher grades of acute GVHD are associated with a higher risk of TA-TMA. 19 An autopsy study by Changsirikulchai et al demonstrated that TA-TMA was 4 times more likely to occur in patients with acute GVHD than in those without. 21 The endothelial injury in such instances was thought to result from circulating cytokines, low levels of vascular endothelial growth factor, activation of the coagulation pathway, or direct endothelial damage from cytotoxic donor T cells. 21
Screening and Evaluation
A renal-centered approach with careful monitoring of renal function (proteinuria, serum creatinine, specific measures of GFR such as cystatin C or Iohexol) 22 can be used. Regular measurement of blood pressure, along with hemogram and lactate dehydrogenase, can be used for screening of TA-TMA 22 (Figure 1). The TA-TMA is suspected in patients who develop the following symptoms without any obvious explanation: a new diagnosis of hypertension requiring >2 antihypertensive medications, proteinuria >30 mg/dL, thrombocytopenia, and features of microangiopathic hemolysis (elevated LDH, the presence of schistocytes, decreased haptoglobin, or increased transfusion needs). 22 In fact, elevated systolic blood pressure has been identified as an early marker of TA-TMA in pediatric patients undergoing HSCT. 24 In a study, patients with TA-TMA were found to have higher average systolic blood pressure compared to non-TA-TMA group. Systolic hypertension was apparent approximately 1 week before the diagnosis of TA-TMA and persisted, despite aggressive antihypertensive therapy. 24
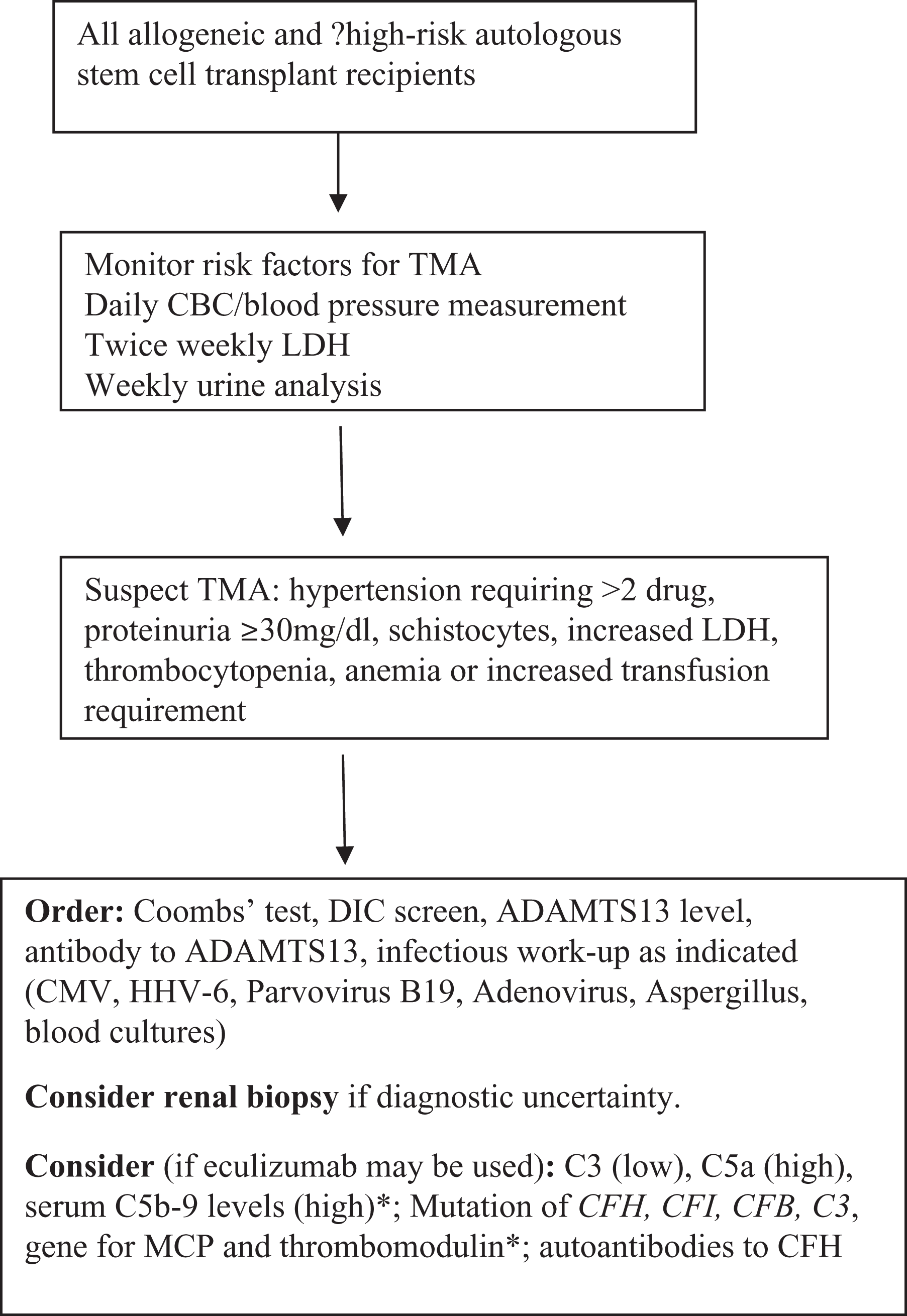
A proposed algorithm for the initial evaluation of hematopoietic stem cell transplant-associated thrombotic microangiopathy. CBC indicates complete blood count; CMV, cytomegalovirus; DIC, disseminated intravascular coagulopathy; GVHD, graft-versus-host disease; HHV, human herpes virus; LDH, lactate dehydrogenase; MCP, membrane cofactor protein; TMA, thrombotic microangiopathy. Early involvement of transplant infectious disease (for possible infection) or nephrology experts (routinely or if renal impairment) is important for optimal management. *see text for details. 2,5,9,15,22,23
The principal manifestations of TA-TMA are similar to those of other conditions such as toxicity from conditioning regimen or other medications, delayed engraftment, infections, acute GVHD, and disease relapse. 10 Therefore, in a patient with possible TA-TMA, the following tests may be performed to diagnose or rule out alternate causes: Coombs test (autoimmune hemolysis may also coexist with TA-TMA), a screening test for disseminated intravascular coagulopathy (eg, fibrinogen, antithrombin activity, prothrombin time, and activated partial thromboplastin time), ADAMTS13 activity level, antibody to ADAMTS13, and infectious evaluation (CMV, herpes viral panel, parvovirus B19, adenovirus, Aspergillus, 2 and blood cultures if there is any concern for sepsis). 1
Select patients with confirmed diagnosis of TA-TMA should be tested for autoantibody against CFH, complement mutation including mutations in CFHR3-CFHR1 genes, complement factor I (CFI), complement factor B (CFB), complement C3 gene, thrombomodulin (THBD) gene, or gene for membrane cofactor protein. 15 Antibody to CFH should be tested in patients’ plasma after the diagnosis of TA-TMA; however, whether recipient, donor, or both genotypes should be assessed for complement mutations is not well established. 15 Until further data are available, assessment of both genotypes may be reasonable and can help establish optimal strategy. Since blood sample from recipient is not routinely stored before HSCT, and DNA obtained from recipients’ blood after engraftment will reflect donor complement genes, assessment of recipient genotypes may not be possible in many circumstances. Complement levels such as low level of C3 and high level of C5a and sC5b-9 has been suggested to possibly indicate a state of complement activation (in aHUS); however, the sensitivity and specificity are low. 23 Although renal biopsy may be considered if the diagnosis of TMA is uncertain, the benefit should be weighed against the risk of bleeding complications in patients with thrombocytopenia. 21,25 Nevertheless, renal biopsy, when safe to perform, may provide useful diagnostic and prognostic information. 25 For example, arteriolar C4d deposits seen in kidney biopsies or biopsies from other sites may support the diagnosis of complement activation. 14,26,27 Skin biopsy to identify focal deposition of C3, C3d, C4d, and C5b–9 may also be helpful in making the diagnosis. 9
Diagnosis
The diagnosis of TA-TMA is complex and requires integration of clinicopathologic data. Although kidneys are prominently affected, involvement of other organs such as the lungs and gastrointestinal tract has been reported. 28 –30 The histologic features of TA-TMA in the kidney include thickened capillary walls, fragmented erythrocytes, occluded vascular lumen, and endothelial cell separation, which is similar to TTP, aHUS, and Shiga toxin-associated HUS 10,30 (Figure 2). Renal biopsy is especially important for diagnosis in the presence of clinical uncertainty or significant renal dysfunction. However, it carries a significant risk of bleeding in many posttransplant patients. 21,25
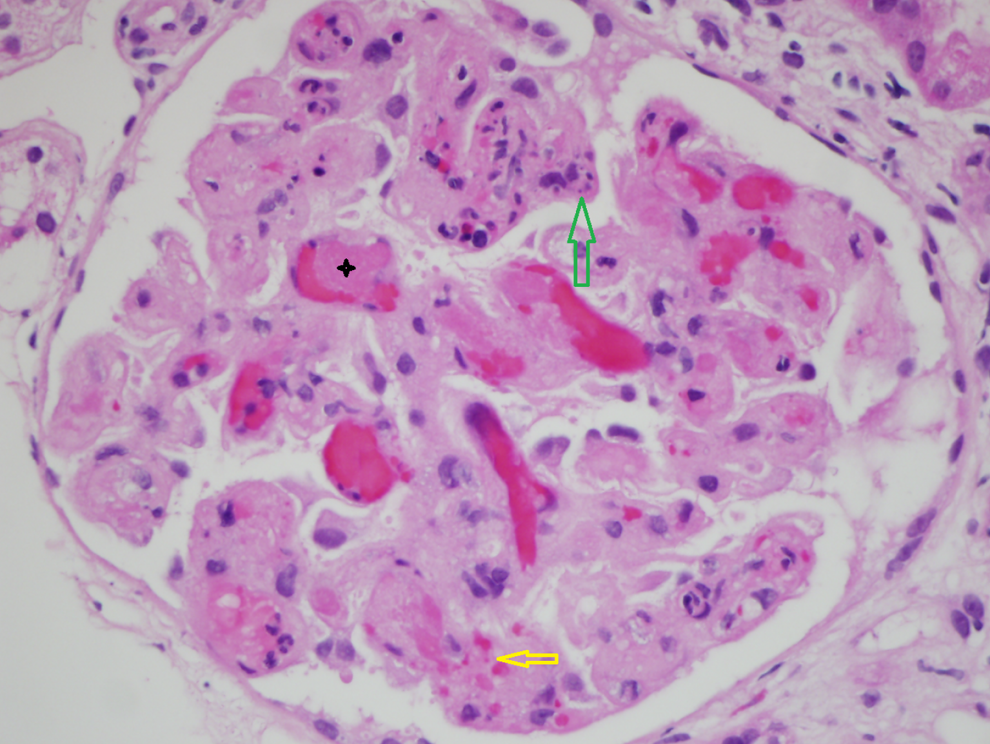
Thrombotic microangopathy involving the kidney. A kidney biopsy (hematoxylin and eosin stain, ×60 magnification) in a patient with chronic graft-versus-host disease demonstrates occlusion of capillary lumina with fibrin thrombi (black star), endothelial cell swelling, red cell fragmentation (yellow arrow), and individual cell necrosis (green arrow).
Since histologic diagnosis is not always possible, several clinical criteria have been proposed for the diagnosis of TA-TMA including a recent set of criteria proposed by Jodele et al 31 (Table 1). Although these criteria provide useful guidelines, the existing criteria are not sensitive and thus may lead to misdiagnosis or delayed diagnosis of TA-TMA. 21,30,33,34 Serum creatinine may be an insensitive marker of renal injury due to reduced muscle mass. 35 The LDH may not be performed routinely in many transplant centers. The presence of schistocytes, which is a nonspecific finding, may not be apparent until several days after clinical symptoms. 36 Further, diagnostic criteria have been criticized for not incorporating uncontrolled hypertension as a possible early marker of TA-TMA and renal dysfunction. 36 In addition to its ease of monitoring, the presence of an elevated blood pressure was identified as an early marker of TA-TMA in a study of pediatric patients with neuroblastoma undergoing autologous HSCT. 24 Proteinuria is another important sign of renal involvement in TA-TMA, which is not included in the existing criteria. 36 Albuminuria after HSCT may indicate renal endothelial injury and may also predict the risk of increased mortality. 37 When biopsy cannot be performed, routine urinalyses every 1 to 2 weeks after SCT may be used to screen for hematuria and proteinuria. 24 Such limitations in the existing criteria and risks of kidney biopsy suggest the need for a high clinical suspicion. Close attention to the renal manifestations of disease, such as new diagnosis of hypertension and proteinuria, may help in early detection of TA-TMA. 35 Better diagnostic tools and identification of markers may allow early diagnosis. 3,25 The new set of criteria proposed by Jodele et al incorporates renal manifestations such as proteinuria, hypertension, and terminal complement activation. 31 Although these criteria were based on prior studies, mainly from their group, the new criteria will need external validation.
Diagnostic Criteria for Stem Cell Transplant Associated Thrombotic Microangiopathy.

Abbreviation: NS, not specified.
aThe diagnosis requires microangiopathy detected on a tissue biopsy or laboratory and clinical markers of transplant-associated thrombotic microangiopathy. Elevated lactate dehydrogenase, proteinuria ≥30 mg/dL and new hypertension may be indicative of transplant-associated thrombotic microangiopathy.
bThrombocytopenia is defined as platelets <50 × 109/L or a >50% decrease in platelet count from baseline.
cRenal dysfunction is described as doubling of serum creatinine or 50% decrease in creatinine clearance from baseline before transplant.
Once a diagnosis of TA-TMA has been made, National Cancer Institute Common Toxicity Criteria adverse event grading system may be used to assess its severity. Grade 1 disease is defined by the presence of schistocytes without clinical findings. Grades 2, 3, and 4 disease additionally have increased creatinine <3 times the upper limit of normal (grade 2), increased creatinine >3 times the upper limit (grade 3), and renal failure requiring dialysis or causing encephalopathy (grade 4). 7 However, the grading system is based on serum creatinine, which is not a reliable marker in patients with HSCT. 35 The identification of more sensitive markers may allow us to refine the severity grading in the future.
Management
Since the underlying mechanisms for TA-TMA is complex and not well understood, the current treatment strategy is suboptimal and associated with poor response rate and survival. Some of the commonly utilized therapeutic options include withdrawal of offending agent such as CNIs, plasma exchange, and rituximab (Figure 3). 3,40,41 In general, patient’s responses to therapy are variable and may arguably depend on the timing of initiation of therapy. 10,18,40 –42 Case series have indicated a possible role of daclizumab, 43 defibrotide, 44 and vincristine. 45,46 More recently, eculizumab has been utilized to manage these patients 13 based on its efficacy in aHUS.
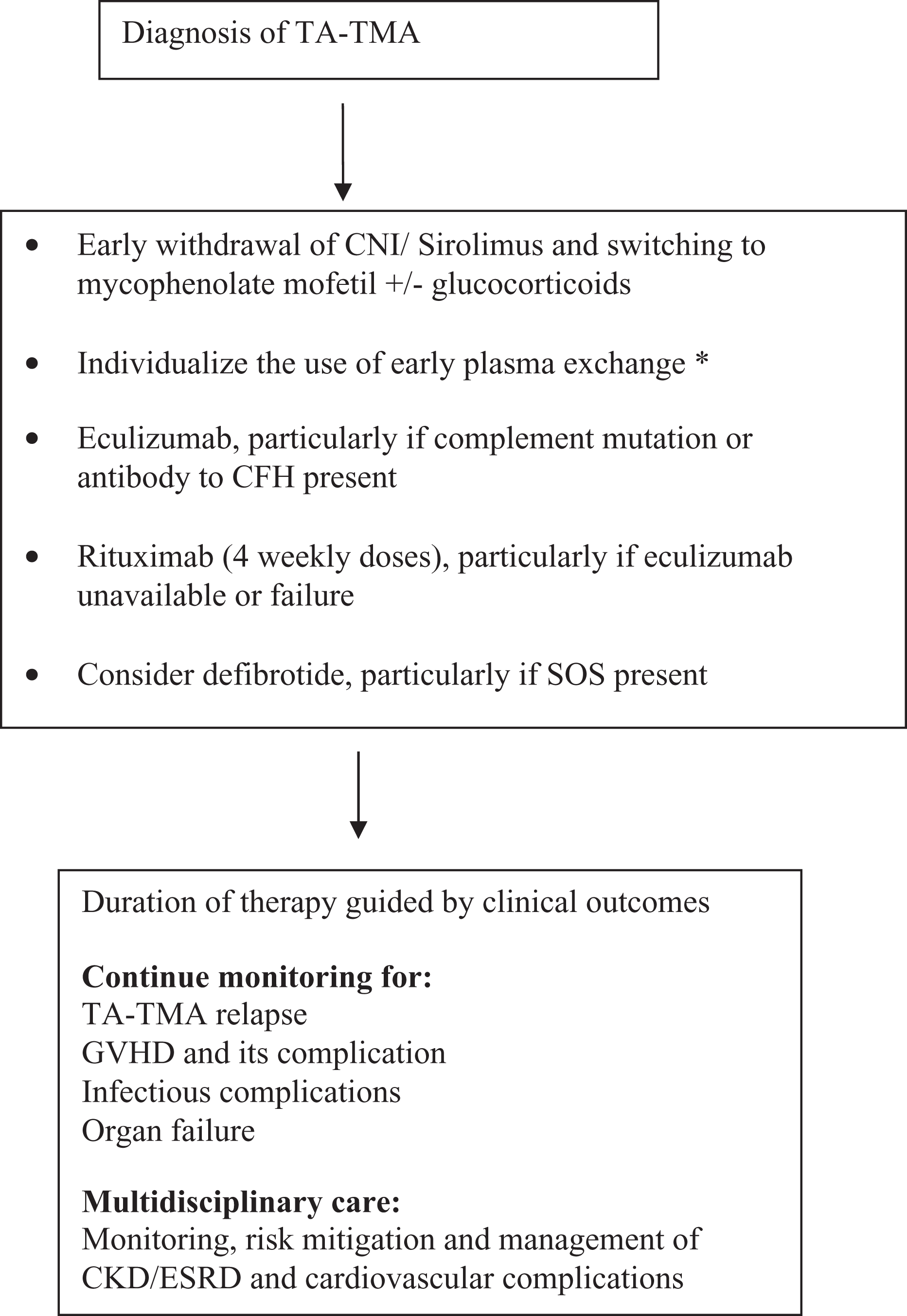
A proposed algorithm for the management of hematopoietic stem cell transplant-associated thrombotic microangiopathy. CKD indicates chronic kidney disease; CNI, calcineurin inhibitor; ESRD, end-stage renal disease; GVHD, graft-versus-host disease; SOS, sinusoidal obstruction syndrome; TA-TMA, transplant-associated thrombotic microangiopathy. *see text for details. 5,13,38,39
Since CNIs constitute one of the important risk factors, discontinuation of CNIs or conversion to another drug plays a pivotal role in the management of TA-TMA. 9,47 In a study by Worel et al, response rates of 63% were observed when these drugs were withheld, and plasma exchange was rapidly instituted. 38 However, discontinuation of CNIs has a potential risk of increasing the risk of GVHD. Replacing CNIs with other immunosuppressive agents may be more beneficial than dose reduction. 7,40,48 Switching to tacrolimus from cyclosporine may be less likely to offer benefit, although some studies have reported successful use of cyclosporine in TA-TMA associated with tacrolimus. 49 –53 Since sirolimus is also associated with the risk of TA-TMA, 48,54,55 whether switching CNIs to sirolimus is a reasonable strategy is unclear. Mycophenolate mofetil with or without corticosteroids may be a better alternative for prophylaxis and treatment of GVHD. 56 Interleukin 2 receptor antagonists such as basiliximab and daclizumab have also shown benefit in a few patients with TA-TMA. 40,43 In an Italian study of 12 patients, the early use of defibrotide resulted in a response rate of 50%. 44
Plasma exchange has been used for several decades to treat TMA including TA-TMA. Although it may benefit temporarily in TA-TMA with an acute response, a large number of reports and case series suggest that it is not effective in the long term. 57 –61 A few authors have raised the possibility that early initiation of plasma exchange may be beneficial; however, given the quality of data, these speculations are at best hypothesis generating. 59 In the Worel study, a high response rate of 63% was noted with CNI withdrawal and initiation of plasma exchange; however, the survival data are not available. Whether CNI withdrawal or plasma exchange played a more important role cannot be separated. 38 A review reported the overall mortality rate of patients with TA-TMA managed with plasma exchange to be about 80%, despite a median response rate of up to 50%. Responses are defined as resolution of laboratory and clinical manifestations of TMA with or without persistent renal impairment. 7 The primary etiology of TA-TMA is not ADAMTS13 deficiency, which may explain why plasma exchange is not as effective in TA-TMA compared to idiopathic TTP. 10 The TA-TMA may perhaps be expected to respond to plasma exchange if ADAMTS13 level is low with activity less than 10% (which is generally not the case) or antibodies against CFH are present. 9
Plasma exchange may also have additional putative mechanisms such as replacement of defective complement proteins or removal of inflammatory cytokines or circulating endothelial cells 5 ; however, this remains to be proven.
Rituximab is an anti-CD20 monoclonal antibody with immunomodulatory effects. In acquired TTP, rituximab can prevent the formation of antibody against ADAMTS13. Hence, it is frequently utilized in TTP, particularly for relapsed or refractory TTP. 1 Because of its role in TTP, rituximab has been used in a few patients with TA-TMA as monotherapy or in combination with plasma exchange. 39,62,63 In a retrospective study by Au et al, 4 of 5 patients with TA-TMA following an allogeneic HSCT were successfully managed with rituximab. At 10 months, 3 patients remained in remission, and the fourth patient succumbed to sepsis. 39 Jodele et al described a response rate of 67% to rituximab with or without plasma exchange in 6 pediatric patients with TA-TMA. 14 Rituximab is speculated to alter immune regulation, antibody production, or complement activation in TA-TMA. 39 There are no guidelines regarding the duration of therapy, which is based on clinical improvement. The commonly used regimen is weekly rituximab (375 mg/m2) for 4 doses.
More recently, eculizumab has been utilized in TA-TMA. Eculizumab is a humanized monoclonal immunoglobulin G antibody that binds to complement protein C5, and thus prevents formation of membrane attack complex and complement-mediated TMA. 64 Since complement dysregulation is important in the pathogenesis of TA-TMA, eculizumab has also been used in few patients with good outcomes. 13 A review of published reports 65 demonstrates the use of eculizumab in a total of 26 cases of TA-TMA following HSCT or solid organ transplant. 9,13,66 –75 The cases were initially treated with discontinuation or dose reduction in CNI with or without plasma exchange. Of these patients, 24 (92%) were alive at 1-year follow-up, 65 and the remaining 2 (8%) pediatric patients did not achieve therapeutic levels even after dose escalation and died. 13
Eculizumab dosage in TA-TMA has largely followed the dosing used in aHUS with induction phase (900 mg weekly for 4 weeks) and maintenance phase (1200 mg for first week, then 1200 mg every 2 weeks). 76 In children, the recommended dose varies from 300 mg for patients weighing 5 to 10 kg to 900 mg for patients weighing 40 kg or more. However, in a pediatric study, patients with HSCT appeared to require higher doses or frequency to achieve a therapeutic level and clinical response. 13 In the initial study of aHUS, a supplemental dose of 600 mg was given before plasma infusion or within 1 hour after plasma exchange; however, in clinical practice, eculizumab monotherapy without plasma exchange is used. Upon initiation of eculizumab, plasma exchange should be avoided in TA-TMA because of concerns about lack of efficacy and removal of eculizumab with plasma exchange. Plasma exchange or plasma infusion (large-volume platelet transfusion also contain plasma) may also reduce eculizumab blood levels by providing exogenous complement factors. In TA-TMA, although the duration of treatment is still uncertain, eculizumab may be stopped after resolution of features of TA-TMA. In a report by Okano et al, a total of 14 doses of eculizumab were used till TA-TMA symptoms resolved. 72 Jodele et al reported a median use of 5.5 doses (range 2-21) in patients who responded to treatment. 13 Eculizumab therapy is associated with an increased susceptibility to encapsulated organisms, so meningococcal vaccine is given if appropriate or antibiotic prophylaxis with ciprofloxacin or penicillin V may be used. 64,77 The TA-TMA occurs mostly during the first 100 days of HSCT, 2 –6 during when HSCT recipients may not mount a response to vaccines. The timeline since HSCT may, therefore, dictate a choice of meningococcal prophylaxis.
Response to any of the above-mentioned therapy may be indicated by improvement in clinical symptoms, increase in platelet counts, and decrease in hemolytic markers. Although kidney dysfunction and proteinuria may resolve in some patients, others may develop chronic kidney disease. Patients, who develop TA-TMA, are also at an increased risk of GVHD, cardiovascular, and other complications. 5,22 Hence, a well-established survivorship plan is important for early detection of such complications and to improve long-term outcomes.
Prognosis
The TA-TMA has poor prognosis with mortality rate as high as 50% to 60%, despite treatment. 2 –6,10,13 The high mortality may be due to direct complications of TA-TMA such as renal failure or due to complications of acute GVHD and infection. Some poor prognostic factors in TA-TMA include age more than 18 years, the use of unrelated or haploidentical donor, elevated TMA index (LDH–platelet ratio), schistocyte count (>5-10/HPF), development of nephropathy, and TA-TMA in the absence of sirolimus exposure. 6,18,48,78 The absence of renal dysfunction predicts a better overall survival. 9
Conclusion
The TA-TMA is a relatively common and serious condition requiring prompt diagnosis, supportive care, and specific treatment. However, a lack of specific diagnostic tests often results in diagnosis late in the disease progression. Besides, current therapy for TA-TMA is still empiric and largely ineffective. Recent advances include better understanding of the underlying pathogenesis including elucidation of the role of complement activation. 13 Recent studies have demonstrated the utility of monitoring of blood pressure, LDH, and urine protein in early detection of TA-TMA. 35 Although the role of plasma exchange is limited, recent reports have indicated a benefit with the use of rituximab. 39,62,63 Early discontinuation of CNIs may also play an important role. 9 Eculizumab has shown promising results but needs to be investigated further. Further research in the molecular pathogenesis, screening for TA-TMA, early discontinuation of CNIs, and use of rituximab or eculizumab in select patients may improve outcomes.
Footnotes
Authors’ Note
Osama Elsallabi and Vijaya Raj Bhatt contributed equally and share the first authorship.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the University of Nebraska Medical Center, College of Medicine, Physician-Scientist Training Program Grant 2015-2016.