
Abstract
This study presents the demographics, clinical spectrum, and outcome of patients with congenital factor VII (FVII) deficiency at a tertiary care center over a period of 12 years. Of the 49 patients, 27 (55%) patients were males. Consanguinity was found in 92% of the patients. The median age of symptom onset was 2.4 (interquartile range [IQR]: 1.1-6.5) years with a median age of 5.8 (IQR: 3.1-10) years at diagnosis. Life-threatening complications like intracranial bleeding (ICB) and intra-abdominal bleeding (IAB) were observed in 8 (16.4%) patients. We found that 11 (55%) of the 20 patients with FVII coagulant activity (FVIIc) <1% were either asymptomatic or showed mild phenotype. In contrast, 9 (53%) of the 17 patients with FVIIc >5% were affected by severe symptoms. Age <1 year was the only identified risk factor associated with development of life-threatening bleeding episodes (P = .042; odds ratio 6.46). Overall, 4 (8.2%) died as a consequence of ICB (3 patients) and IAB (1 patient).
Introduction
Congenital factor VII (FVII) deficiency (Online Mendelian Inheritance in Man: 227500) is the commonest among the rare congenital coagulation disorders and is characterized by autosomal recessive inheritance. 1 Clinically symptomatic FVII deficiency has an estimated prevalence of 1:500 000 without any racial/ethnic predilection. 2 However, true prevalence of FVII deficiency is probably higher given the presence of asymptomatic and mildly symptomatic individuals. 3 A procoagulant, FVII, plays a pivotal role in the extrinsic coagulation pathway. Its activated form VIIa combines with tissue factor, released from damaged endothelial cells causing factor X activation which through a series of events leads to the formation of a stable, fibrin clot. Additionally, FVII augments intrinsic pathway by activating factor IX. 4,5
Clinical heterogeneity is a feature of this hemorrhagic disorder concerning both sites and severity of bleeding that does not always correlate with the level of FVII coagulant activity (FVIIc) measured in plasma. 6 A disease severity classification has been formulated based on the timing and clinical presentation of the disease. 3 The International Registry Factor Seven Study Group (IRF7SG) classifies bleeding episodes as severe, moderate, or mild (Table 1). 7 Most severe cases of FVII deficiency are diagnosed during childhood, often during the first 6 months of life. In infancy, the gastrointestinal tract and central nervous system (CNS) are the most common sites, accounting for 60% to 70% of bleeds in this age group. 8,9 In mildly and moderately affected patients, symptoms are minimal and often restricted to post-traumatic/surgical hemorrhage. 3
The Clinical Severity Classification Recommended by the IRF7SG. 7

Abbreviations: CNS, central nervous system; GI, gastrointestinal; IRF7SG, International Registry Factor Seven Study Group.
The laboratory diagnosis of FVII deficiency is suggested by a prolonged prothrombin time (PT) with a normal activated partial thromboplastin time (aPTT) and confirmed based on a low FVII assay. However, being a rare bleeding disorder, it needs a high index of suspicion. Molecular diagnosis is available, and a broad spectrum of mutations has been characterized in the FVII gene that is located on chromosome 13. 4,5 Molecular analysis of a small series of 10 FVII-deficient patients found 5 novel mutations in Pakistan. 10 This heterogeneity in the genetic mutations may explain the broad spectrum of clinical presentations seen to be associated with FVII deficiency.
A number of studies have been conducted in the past, with data obtained from multiple centers and blood banks that provide a point prevalence of a number of inherited coagulation disorders with common clinical presentations. 1,2,11,12 However, there have not been any studies focusing solely on the demographics, the implications of consanguineous marriages, clinical presentations, fatal complications, and long-term effects of FVII deficiency in the population. The aim of this study was to describe the baseline demographics, clinical spectrum, hemostatic values, and outcome of 49 pediatric patients who were diagnosed with congenital FVII deficiency in a tertiary health care facility of Karachi, Pakistan.
Methodology
This is a retrospective review of patients aged 0 to 18 years who were diagnosed with FVII deficiency, at Aga Khan University Hospital (AKUH), Karachi, over a period of 12 years from January 2001 to December 2012.
Data Collection
Admitted patients were identified using coded discharge records with the diagnosis of coagulation/factor deficiency, as there is no specific international classification of disease (ICD) code available for FVII deficiency, ICDs (2008; code 286.3) were used for the identification of inpatient study populations. Outpatients were identified by outpatient department record. Computerized laboratory system was utilized as an additional source for identifying FVII-deficient patients. All the FVII-deficient patients were diagnosed on the basis of having a minimum number of low FVII (<50%) levels. Patients’ medical charts were reviewed to evaluate nature of FVII deficiency—inherited or acquired. Patients with acquired FVII deficiency secondary to liver disease, vitamin K deficiency, or malabsorption were identified through prolonged PT and aPTT as well as through multiple factor deficiencies and were excluded from the study. These details were also verified through laboratory information system using medical record number for each patient.
Laboratory Workup
All tests were performed at the coagulation laboratory of The Aga Khan University Hospital which is an ISO 9000 and Joint Commission International-accredited laboratory. Besides stringent internal control, it regularly participates in external quality assessment by College of American Pathologists.
Briefly, 1.8 mL of blood was collected in 3.2% sodium citrate (BD, Becton Dickinson and Company, New Jersey). Prothrombin time and aPTT were performed on Sysmex CA-1500 system (Sysmex Corporation, Kobe, Japan) using Dade, Innovin, and Actin FS reagents. 13 The instrument and reagents remained unchanged during the study period. Reference range established in our laboratory for FVII ranged from 50% to 150%. The diagnosis of FVII deficiency was typically considered on the discordance between the PT and the normality of the aPTT. Factor VII assay was performed on the same analyzer as described earlier utilizing clotting-based methodology. All abnormal tests were repeated to confirm the results, and the most recent level was used for stratification of severity of FVII deficiency. Those having isolated FVII deficiency were evaluated for correction of PT with normal plasma in a ratio of 1:1 (PT mixing test). The patients in whom this could not be achieved were considered to have acquired autoimmune FVII deficiency and were also excluded from the study.
Data Handling
Data were collected on a prestructured proforma by reviewing medical records and phone interviews that were done to complete the missing data by primary investigator. Information including gender, age at presentation and at the time of diagnosis, presenting complaints, family history, consanguinity, applied treatment, surgical interventions, complications, and follow-up visits were recorded. The patients were also evaluated for type, site, frequency, nature, and severity of bleeding manifestations. Hemoglobin level, platelet count, peripheral smear, and result of coagulation screen (PT, aPTT, and BT) were also recorded. Patients were grouped according to clinical severity classification (Table 1) as recommended by the IRF7SG 5 and FVIIc level for further analysis.
Statistical Analysis
For data entry and analysis, SPSS version 20.0 (IBM, Chicago) was used. Median with interquartile range (IQR) of age and mean with standard deviation of FVII levels was computed, while frequency (percentage) for gender, consanguinity in parents, family history of bleeding disorder/FVII deficiency, and complications were calculated. Regression analysis was performed for the identification of risk factors for life-threatening bleeding episodes, and crude odd ratio (OR) was calculated. A P value of <.05 was considered significant at the univariate level.
Ethical Approval
The study was approved by the Ethical Review Board (ERB) of Aga Khan University, Karachi (2396-Ped-ERC-12).
Results
Demographics
A total of 49 FVII-deficient patients were identified during the study period. Table 2 describes the demographic features of all enrolled patients. There were 27 (55%) males and 22 (45%) females. The rate of consanguinity among the parents of patients was 92%. In all, 5 patients had bleeding disorder in their siblings, while significant family history of bleeding was observed in 7 patients.
Demographic Parameters of Factor VII-Deficient Patients.
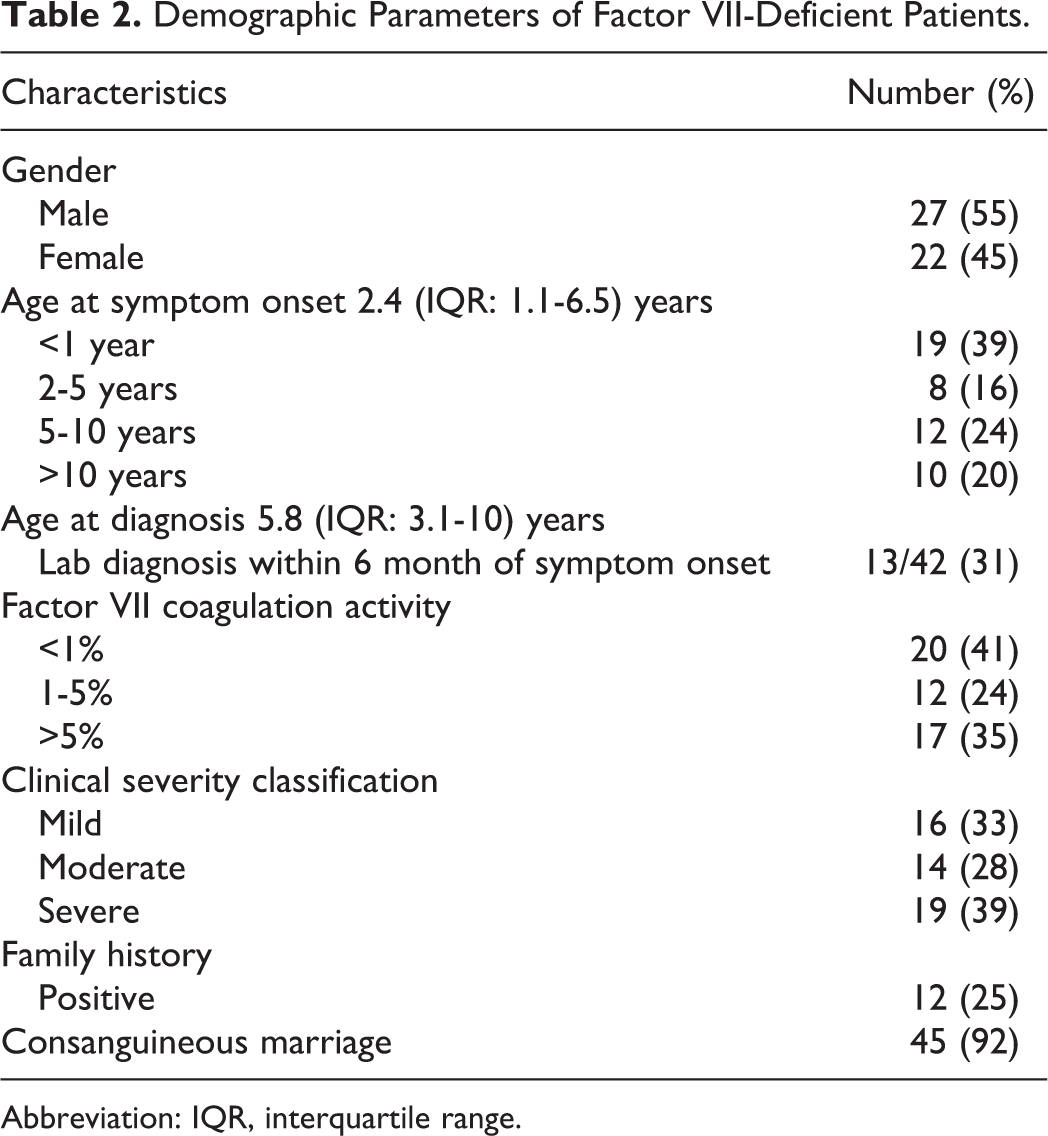
Abbreviation: IQR, interquartile range.
Age of Onset and at Diagnosis
The median age of symptom onset of this cohort was 2.4 (IQR: 1.1-6.5) years and median age at the time of diagnosis was 5.8 (IQR: 3.1-10) years. Age at symptom onset was compared with age at laboratory diagnosis in the 42 patients for whom both time points were available. In only 31% (13 of 42) of the patients, diagnosis of FVII deficiency was made simultaneously or within 6 months of symptom onset. There was considerable delay in establishing laboratory diagnosis in the remaining patients, median age at diagnosis of this cohort (29 of 42) was 6.9 (IQR: 4.1-11) years, and mean time lag before laboratory diagnosis was 4.3 (IQR: 1.8-9.5) years.
Symptomatology
The bleeding sites and frequency of bleeding episodes are shown in Figure 1. The most common symptoms were epistaxis, easy bruisability, prolonged umbilical cord and post-circumcision bleeding, gastrointestinal (GI) bleeding, and gum bleeding. Intracranial bleeding (ICB) and intra-abdominal bleeding (IAB) were among the life-threatening events and were observed in 8 (16.4%) patients. Comparison of the mean age at presentation for specific symptoms yielded a significant difference. In particular, of the 8 patients who presented with life-threatening bleeding, 6 (75%) of them presented within the first year of life. In our study, all the bleeding episodes were treated with fresh frozen plasma (FFP) transfusions. Additionally, in 3 cases of impending life-threatening bleeding recombinant-activated FVII (rFVIIa 70-90 μg/kg) was given before surgery. All patients were then kept on regular FFP transfusion until the hemostasis was achieved.
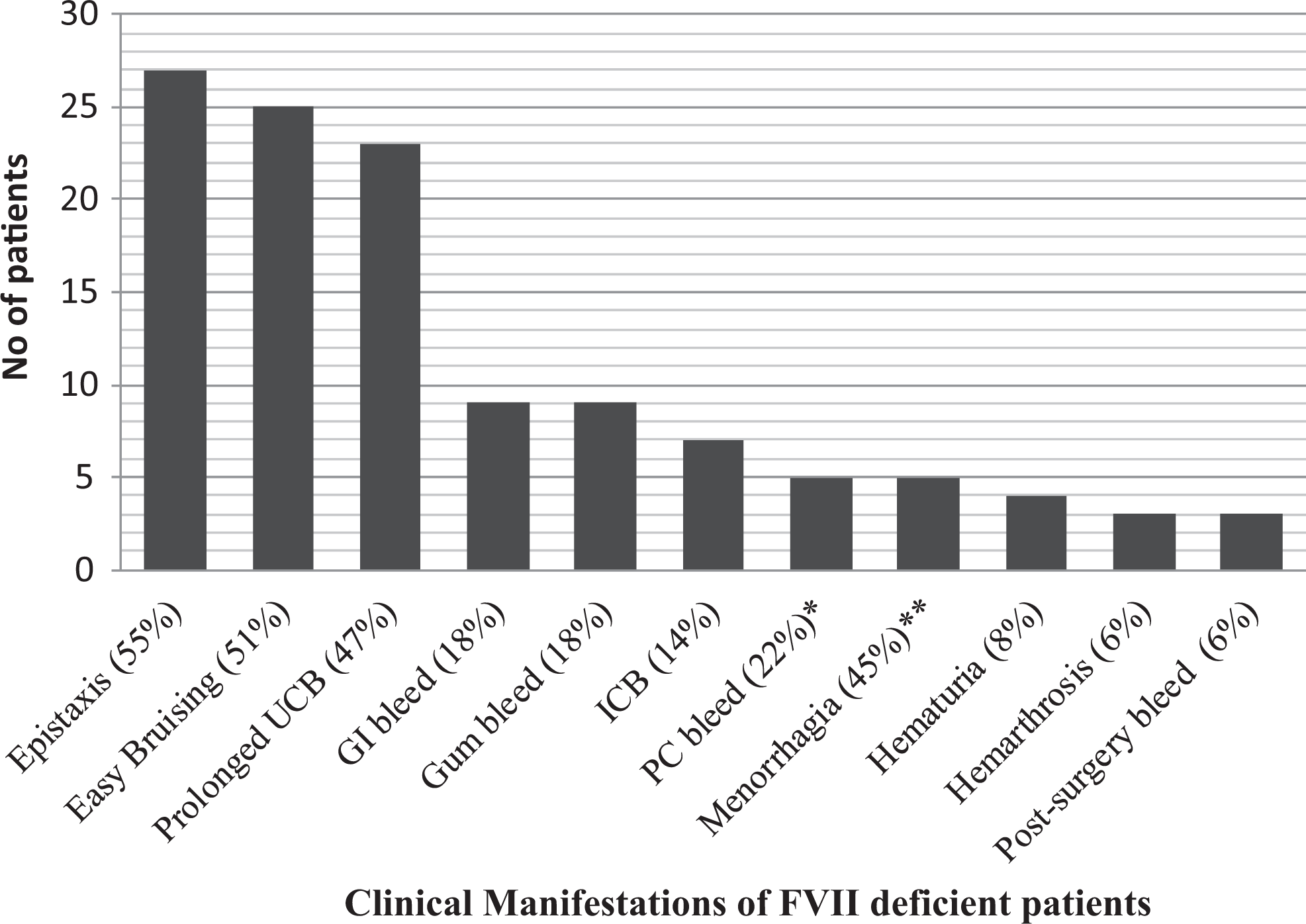
The bleeding sites and frequency of bleeding episodes of symptomatic patients. ICB indicates intracranial bleed; GI, gastrointestinal; UCB, umbilical cord bleed; PC bleed, post-circumcision bleed. *Sex adjusted frequency for PC bleed is 22% (6 of 22). **Age and gender adjusted frequency for menorrhagia is 45% (5 of 11).
Severity, FVII level, and Complications
According to clinical severity classification, 16 (33%), 14 (28%), and 19 (39%) patients had mild, moderate, and severe phenotype, respectively. The levels of FVIIc were as follows: 20 (41%) patients; <1%, 12 (24%) patients; 1% to 5%, and 17 (35%) patients; >5% to 50%. We found that 11 (55%) of the 20 patients with FVIIc level less than 1% were either asymptomatic or showed a very mild phenotype. In contrast, 9 (53%) of the 17 patients with FVIIc level above 5% were affected by severe symptoms (GI in 4 patients, CNS in 3 patients, and hemarthrosis in 2 patients). A total of 8 patients had life-threatening bleeding episodes (7 ICB and 1 IAB bleedings), 4 of whom died as a consequence of bleeding.
Outcomes
Of the 4 (8.2%) mortalities, 3 patients died due to ICB and 1 due to IAB. Age less than 1 year (P = .042; crude OR 6.46) was the only identified risk factor associated with the development of life-threatening bleeding episodes on regression analysis that compared the FVII-deficient patient with and without life-threatening bleeding episodes (Table 3). Of the 45 alive patients, 22 patients have no further bleeding complaints, 15 have on and off minor, self-resolving bleeding episodes, and 9 patients are on regular monthly prophylactic FFP transfusions and doing well with no major bleeding episodes per telephonic interviews. The rationale behind the monthly FFP transfusions could not be ascertained as the interviews were with the patient’s families and not the treating physicians. Outcome data for the remaining 3 patients were missing because they lost to follow-up.
Comparison of Factor VII-Deficient Patient With and Without Life-Threatening Bleeding Episodes.
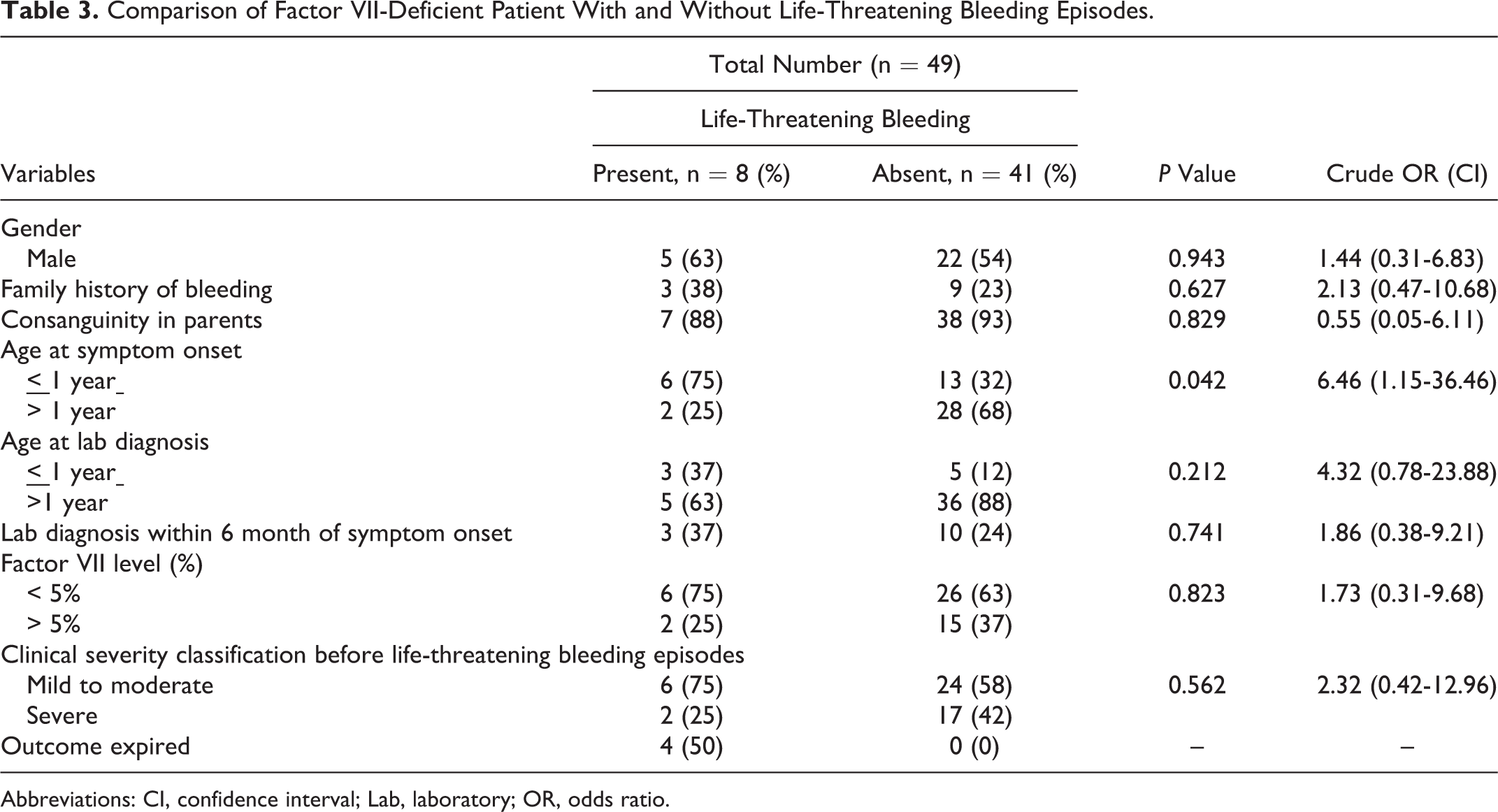
Abbreviations: CI, confidence interval; Lab, laboratory; OR, odds ratio.
Discussion
Many centers and study groups are evaluating data for rare congenital factor deficiencies for a better understanding of their genetic characteristics, clinical presentations, and developing treatment protocols since the 2000s. 11,12 The World Federation of Hemophilia and Rare Bleeding Disorders Database are among some examples of such initiative. 14 Others include the International Registry on Congenital Factor VII deficiency and the Seven Treatment Evaluation Registry.
Our single-center study group comprised 49 pediatric patients diagnosed with FVII deficiency over a period of 12 years with a median age of 5.8 (IQR: 3.1-10) years at diagnosis. The minimum age of presentation ranged from 0 years to a maximum age of 18. Although this congenital deficiency is rare globally, our single study experience revealed a large number of FVII-deficient patients that can be attributed to a high frequency of consanguineous marriages in the region and to the autosomal recessive pattern of inheritance of the disease. As with most autosomal recessive disorders, incidence of congenital FVII deficiency is directly associated with consanguinity in parents. Hussain and Bittles reported that approximately 60% of all Pakistani marriages were consanguineous and 80% of these are between first cousins. 15 Therefore, diseases with familial tendencies may be associated with higher prevalence in Pakistan than in other countries.
Literature review revealed a wide age distribution for diagnosis among registered FVII-deficient patients by different group. The IRF7SG study reported an age distribution range of 1 to 90 years, 7 and the mean age of 30 years was reported by Herrmann et al. 16
Studies report that the most common bleeding sites are skin and mucosal tissue, 6,12 most commonly, for example, epistaxis, menorrhagia, gum bleeding, and easy bruising. In our study, prolonged umbilical cord bleeding 32.7% (16 of 49), bruising 30.6% (15 of 49), and epistaxis 26.5% (13/49) were the 3 most common presenting complaints of patients with mild to moderate bleeding. Hemorrhage involving the CNS was seen in 14.3% (7 of 49) of the patients, a figure that matched findings of 16% (12 of 75) and 17% reported by Ragni et al 17 and Peyvandi et al, 12 respectively, whereas the IRF7SG study and Salcioglu et al 18 reported the frequency of CNS bleeding as 7% and 19% (8 of 42), respectively.
Intracranial bleeding is known to be a life-threatening complication and a significant risk of early mortality in deficient individuals. 19 This was reiterated by our study, in which 3 (6.1%) study participants died due to ICB. Hemarthrosis was a less common presentation in our study (6%) as opposed to other literature, which showed a bleeding frequency of 16.6% to 22%. 3,12 Children less than 5 years were most commonly affected, especially as this is the time when they start to crawl and walk. 20 The frequency of gastrointestinal bleedings, 14.3% in our study, was comparable with figures of 14% reported by Peyvandi, Lapecorella, and IRF7SG.
Many studies report menorrhagia as a presenting complaint in females with FVII deficiency. Prevalence is shown to be as high as 60%. 21,22 The frequency of menorrhagia in FVII-deficient individuals is in accordance with the findings in other bleeding disorders like von Willebrand disease. 8 Our study, however, only indicates 10% prevalence of menorrhagia in girls with FVII deficiency. This discrepancy is most likely due to the fact that majority female participants in our study were of a younger age group and had not attained menarche yet. In our study, however, age and sex-adjusted frequency for menorrhagia was 45% (5 of 11).
Bleeding in the severest of cases occurs in the first 6 months of life, but it is not uncommon to observe spontaneous bleeding and bleeding after provocation, such as during and after surgery in older age groups. One study showed that as high as 76% of FVII-deficient patients were diagnosed in their first year of life. 18 It has also been seen that infants and neonates with low-FVII levels carry a heavy bleeding risk. 5
Also, it was seen that there was a delay in diagnosis of the deficiency. Our study showed that the mean age of symptom onset was seen to be significantly different from the age of diagnosis. This observation could be explained by the fact that patients are often treated with packed red cells and various other forms of blood products that tend to mask the underlying coagulopathy. The need for diagnostic investigations instead of just symptomatic treatment is highlighted by this finding.
It is not rare to see a lack of bleeding complications, either spontaneous or provoked, in those with circulating functional factor levels of less than 1%. 23 –25 On the other hand, there have been reports of severe bleeding in patients with factor levels of more than 5%. 26 Likewise, our study shows that 55% of the patients with FVIIc <1% were either asymptomatic or mildly symptomatic, whereas 53% patients with FVIIc >5% were affected by severe symptoms involving the GI system, CNS, and joints.
The life-threatening nature of FVII deficiency also came to light in our study. There were 4 (8.2%) deaths, 3 of which was a result of ICB and 1 of which was secondary to an IAB. Intracranial bleeding and IAB are serious complications and other studies also identify them as significant contributors to morbidity. 2 Studies on boys with hemophilia show that intracranial hemorrhages, even when adequately treated, have poor long-term outcomes, especially in the domain of quality of life. 27
As with other factor deficiencies, substitutional therapy is the main stay of treatment in FVII-deficient individuals with a history of bleeding. Asymptomatic patients undergoing minor surgical procedures can be successfully treated with Tranexamic acid. Prophylaxis is not a common practice; however, reports suggest that prophylaxis may be beneficial in unweaned infants, menorrhagia with iron deficiency, and recurrent haemarthrosis. 28,29 Moreover, continuous infusion of rFVIIa in surgical procedures has also been reported. Complications of treatment are rare and include transmission of blood-borne viruses, thrombosis, and inhibitors to FVII. 30
Limitations of this study include those inherent to its single center, retrospective design particularly selection bias and information bias. Additionally, we do not have the facilities for detection of specific mutations in these patients, and relationship between genotype and phenotype remains to be investigated in future.
Conclusions
Clinical manifestations are variable and do not necessarily correlate with factor levels but its deficiency is associated with severe bleeding and death, particularly in the infant and neonatal proportion of the pediatric population. Therefore, it is important for neonatologists and pediatricians to investigate bleeding diathesis in this age group thoroughly. With a high rate of consanguinity and subsequently higher rate of coagulation deficiencies in the region, large-scale multicenter studies are warranted to assess the magnanimity of the situation.
Footnotes
Authors’ Note
MMA designed the study, performed statistical analysis, and wrote the article accordingly. KAR and PJ wrote the clinical part of the article. ZF and BM helped in designing the study and critically reviewed the article. The authors stated that they had no interests which might be perceived as posing a conflict or bias.
Acknowledgment
The authors gratefully thank Scheherzade Aslam for helping in data collection.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.