
Abstract
Congenital factor VII deficiency is the most common form of rare coagulation factor deficiencies. This article presents a retrospective evaluation of 73 factor VII deficiency cases that had been followed at our center. The study consisted of 48 males and 25 females (2 months-19 years). Thirty-one (42.5%) of them were asymptomatic. Out of symptomatic patients, 17 had severe clinical symptoms, whereas 8 presented with moderate and 17 with mild symptoms. The symptoms listed in order of frequency were as follows: epistaxis, petechia or ecchymose, easy bruising, and oral cavity bleeding. The genotype was determined in 8 patients. Recombinant activated factor VII (rFVIIa) was used to treat 49 bleeding episodes in 8 patients after 2002. In 2 patients with repeated central nervous system bleeding prophylaxis with rFVIIa was administered. No allergic and thrombotic events were observed during both treatment and prophylaxis courses. Antibody occurrence was not detected in the patients during treatment.
Introduction
Congenital factor VII (FVII) deficiency is a rare autosomal recessive disease encountered at a rate of 1:500 000. It was first defined in 1951 by Alexander et al. 1 By activating FIX and FX in the early phase of coagulation, FVII plays a significant and pivotal role in the initiation of coagulation.2,3 Patients with FVII deficiency are classically grouped as type 1 and type 2. In type 1, there is a decrease in both functional activity and antigen level, whereas in type 2, antigen level is normal or slightly low but a dysfunctional protein is present and factor activity is decreased. 4
Most of the patients with FVII deficiency are diagnosed by prolonged prothrombin time (PT). In these patients, unlike hemophilia A and B, there is a slight correlation between procoagulant activity and both frequency and severity of bleeding episodes. This explains the wide clinical spectrum that ranges from asymptomatic patients to cases with fatal central nervous system (CNS) bleedings. The reason for these heterogeneous clinical features has not been fully understood.5 –7
Starting from the 2000s, many centers and study groups have commenced reviewing the data regarding rare factor deficiencies including FVII deficiency. These efforts are made for understanding the clinical and genetic characteristics of FVII deficiencies, developing treatment protocols, and sharing the results.3,8,9 The World Federation of Haemophilia (WFH) and Rare Bleeding Disorders Database (RBDD) are among the foremost examples of such centers.10,11 In the RBDD context, Spreafico et al had collected data from 19 countries and genetic analyses had been performed in 400 patients. 12 The phenotypes, results of therapeutic applications, and genetic mutations of FVII-deficient patients were collected and also reported by many special registry systems.13 –16
Fresh frozen plasma (FFP), prothrombin-complex concentrate (PCC), activated PCC (aPCC), plasma-derived human FVII (pdFVII), and recombinant activated FVII (rFVIIa) may be used for bleeding episodes in FVII deficiency. Recombinant FVIIa (Novo Nordisk, Denmark) is a recombinant product that can be easily dose adjusted and administered without any serious side effects. For these reasons, its use has been preferred mostly.13,17,18 Two studies published by Mariani and Brenner in the treatment of congenital FVII deficiency with rFVIIa are noteworthy in that aspect.19,20
There is a consensus on the necessity of prophylaxis for hereditary FVII deficiency, when severe bleeding episodes have manifested themselves particularly in the newborn and early breast-feeding period. Experiences regarding prophylaxis with both FFP and rFVIIa were reported in series consisting of small number of young children.19,21 –29
In this article, we aimed to present the largest pediatric patient group series from a single center in Turkey. We also wanted to share our experience with rFVIIa in FVII-deficient patients since 2002.
Patients and Methods
Seventy-three FVII-deficient children were followed between the years 1990 and 2008 at our hematology–oncology clinic. The past medical records of the patients were evaluated retrospectively. The ages of the patients at the date of admission, clinical and laboratory findings, bleeding sites, number of bleeding episodes, applied treatments, and surgical interventions were recorded. Initial tests including blood count, PT, activated partial thromboplastin time (aPTT), fibrinogen, bleeding time, and peripheral blood smears were performed. Determinations of PT were done using a commercial PT reagent (Thromborel S of Dade-Behring/Siemens) and FVII assays were performed as 1-stage clotting-based tests using FVII-deficient plasma on different coagulation analyzers (BCT, BCS of Dade-Behring/Siemens or CA-1500 of Sysmex) depending on availability. FVII:antigen tests of our cases could not be performed. Genetic mutational analyses were performed in only 8 patients. The International Registry Factor Seven Study Group (IRF7SG) recommendations regarding the severity of bleeding episodes were used for the classification of our patients (Table 1 ). 13 The term “phenotype” mentioned in this classification is absolutely a clinical definition and it is independent from FVII:C levels of the patients.
The Classification of Bleeding Episodes According to Severity Recommended by the IRF7SG

Abbreviations: CNS, central nervous system; GI, gastrointestinal.
For managing bleeding episodes of our patients, local interventions, FFP, and antifibrinolytics were used until 2002. After 2002, rFVIIa was used to treat 49 bleeding episodes of our 8 patients. The rFVIIa dose of 15 to 35 mcg/kg was repeated every 4 to 6 hours. The median dose was 25 mcg/kg. After bleeding had been stopped, maintenance doses were administered at 6- to 8-hour intervals depending on the characteristics of the bleeding site.
In the first section of this article, we aimed to report our findings of congenital FVII-deficiency cases whereas in the second section we discussed treatment outcomes of rFVIIa therapy.
Results
The age of the 73 patients at the time of admission was between 2 weeks and 19 years (mean 2.2 years). The patient population consisted of 48 (65.8%) males and 25 (34.2%) females. The rate of consanguinity among the parents of patients was 54.8%. The levels of FVII:C were as follows: 13 (17.8%) patients; <5%, 17 (23.3%) patients; 5% to 30% and 43 (58.9%) patients; 30% to 55%.
Forty-two (57.5%) patients were symptomatic and 31 (42.5%) were asymptomatic. Of the asymptomatic patients, 9 (29%) were female and 22 (71%) were male. FVII:C levels of patients presenting with severe bleedings were ranging between 0% and 29%. This value was 0.1% to 34% for patients with moderate bleeding and ranged between 3% and 55% for patients with mild bleeding. The bleeding sites and frequency of bleeding episodes were shown in Table 2 . The mucosa and skin were the most common bleeding sites, whereas CNS was the site of most severe bleeding. The severity of bleeding according to the age at the time of diagnosis was shown in Table 3 . Seventeen patients applied with severe bleeding and 13 (76.5%) of them presented within the first year of life.
The Bleeding Sites and Frequency of Bleeding Episodes of Symptomatic Patients
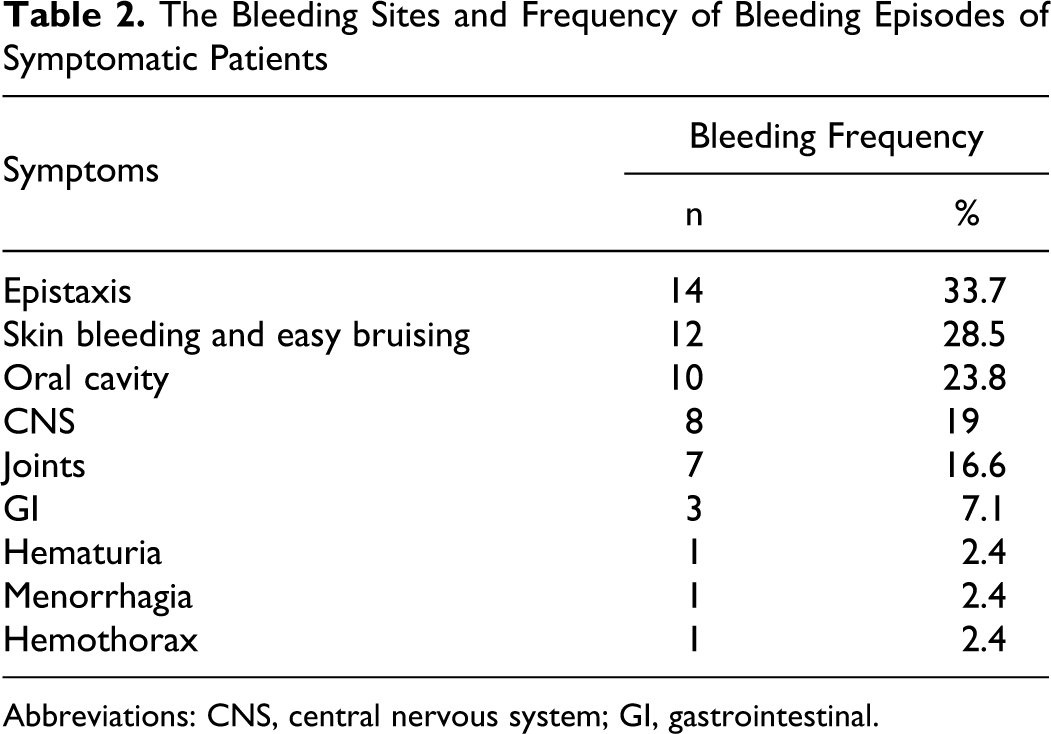
Abbreviations: CNS, central nervous system; GI, gastrointestinal.
Age at the Time of the Diagnosis Were Distributed According to the Severity of Bleeding

The relationship between phenotypes and FVII:C levels of the patients are shown in Table 4 . Eight (47%) out of 17 patients with severe bleedings had FVII:C levels of <5%, and 9 (53%) with levels >5%. The characteristic features of 8 patients with genetic analysis results were reported in Table 5 .
The Relationship Between Phenotypes and FVII:C Levels of the Patients
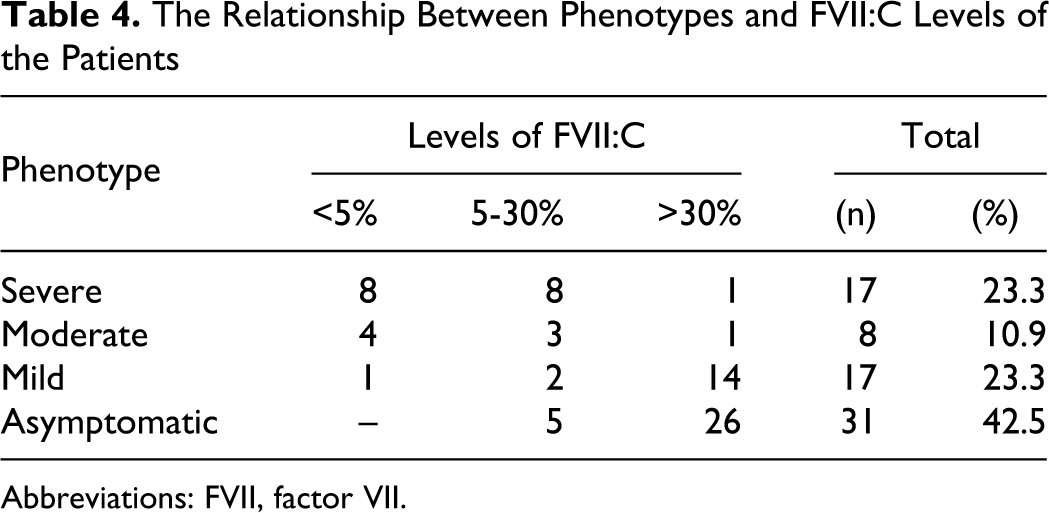
Abbreviations: FVII, factor VII.
The Characteristic Features of Patients in Whom Mutation Analysis Were Performed

Abbreviation: FVII, factor VII.
Fourteen surgical interventions were applied without any bleeding complications. Three of them were major surgical interventions, including ventriculoperitoneal shunt operation, shunt valve replacement, and tonsillectomy. Minor surgical interventions include circumcision (n = 6), tooth extraction (n = 4), and nail extraction (n = 1).
Recombinant FVIIa was used to treat 49 bleeding episodes in 8 patients. General characteristics of patients treated with rFVIIa and the surgical interventions applied were listed in Table 6 . During bleeding episodes and surgical interventions, rFVIIa at doses of 15 to 35 mcg/kg (median 25 mcg/kg) were given every 4 to 6 hours and no higher and additional doses were required. The number of doses had ranged between 1 and 83. Of those, 12 (24.5%) were of severe bleeding episodes, consisting of 7 (58.4%) with CNS bleeding, 2 (16.7%) hemarthrosis, 1 (8.3%) hemothorax, 1 (8.3%) head trauma, and 1 (8.3%) gastrointestinal (GI) bleeding. The sites of bleeding and number of bleeding episodes of the patients who were treated with rFVIIa were presented in Table 7 . No allergic and thrombotic events were observed during treatment and prophylaxis courses. Occurrence of antibody was not observed. None of our patients died due to bleeding.
General Characteristics of Patients in Whom Bleeding Episodes Were Treated With rFVIIa, Were Applied Surgical Interventions, and Were Given Prophylaxis With rFVIIa
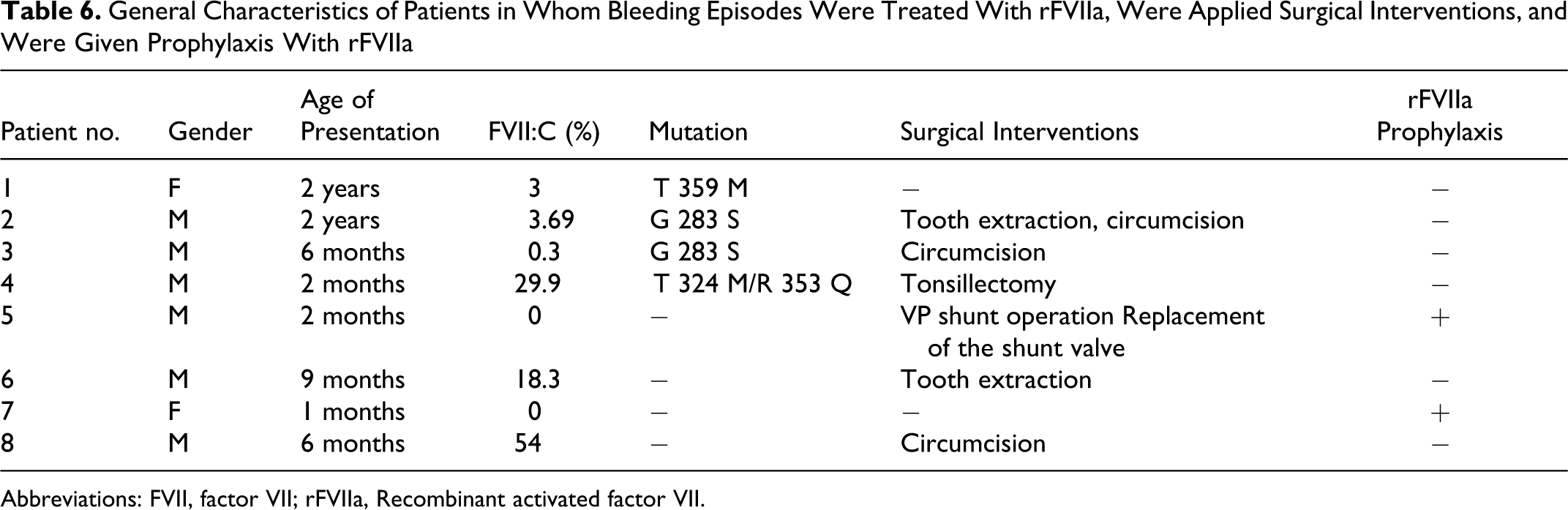
Abbreviations: FVII, factor VII; rFVIIa, Recombinant activated factor VII.
Bleeding Sites and Number of Bleeding Episodes of the Patients Who Were Treated With rFVIIa
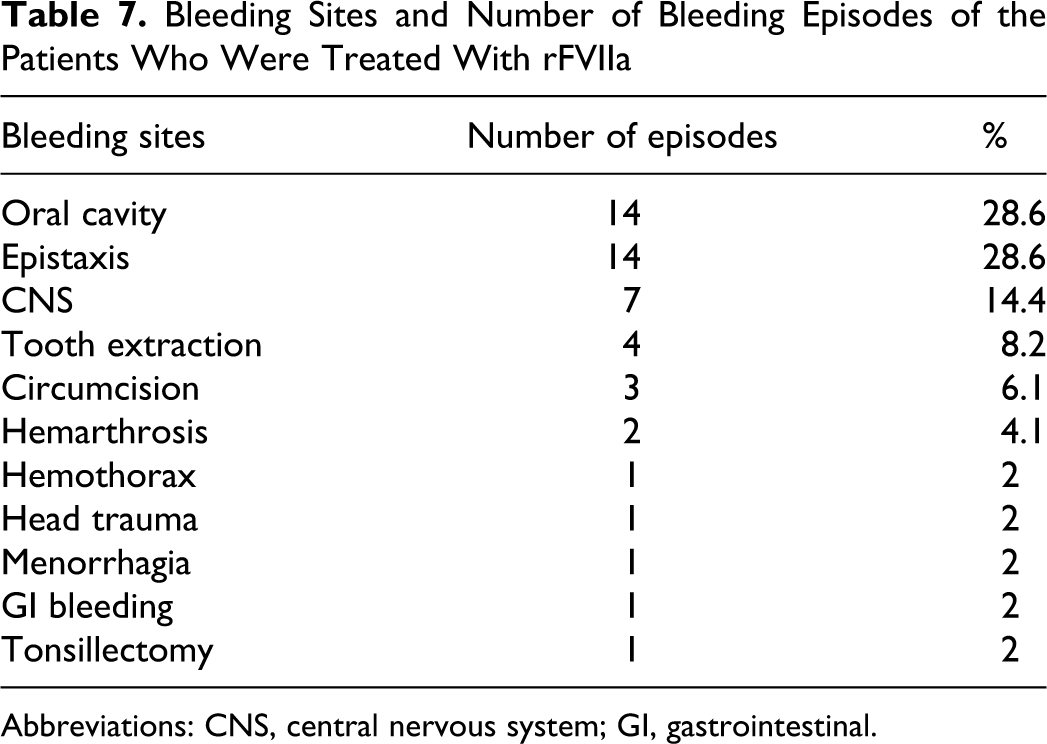
Abbreviations: CNS, central nervous system; GI, gastrointestinal.
Discussion
It is well known that FVII deficiencies constitute the largest group among rare coagulation factor deficiencies with a prevalence of 1:500 000. According to the Turkish Ministry of Health’s 2008 data, out of 361 rare coagulation factor deficiencies, 240 were FVII-deficient patients. 30 Our single-center study group consisted of 73 children and even this will point out to the fact that FVII deficiency is not a rare disorder in our country. The higher frequency of consanguineous marriages (54.8%) that was reported in our study also supports this opinion.
In the literature, large series of FVII deficiencies usually involve both children and adults. Age distribution range was reported as 1 to 90 years in the IRF7SG study, 13 and the mean age was 30 years in the study of Herrmann et al. 16 The ages of our patients when they had presented to our clinic were ranging between 2 weeks and 19 years (mean 2.2 years). Twenty-six percent of them were diagnosed when they were younger than 1 year of age. The dissimilarity in our patients' ages from the previous series may account for the variability in the clinical features and treatment outcomes.
Factor VII deficiency may present with diverse clinical phenotypes ranging from asymptomatic condition to severe disease. Herrmann et al 16 had reported in their study that 71% of homozygous, 50% of double heterozygous, and 19% of heterozygous were found to be clinically asymptomatic. Asymptomatic cases constituted 39.2% of the study group in the IRF7SG trial. 13 Of our cases, 42.5% were asymptomatic and the diagnosis was made either according to family history or by routine laboratory workup prior to a surgical intervention. Seventeen patients had severe phenotypic features and 13 of them (76.5%) were diagnosed in their first year of life. According to our knowledge, there is not any published data having such a high rate of severe bleeding case series before first age. Although severity of the bleedings may not directly be associated with FVII:C levels, newborns and early infants with low FVII:C levels carry a heavy bleeding risk. 13
The most frequently reported bleeding sites are skin and mucosal tissue.14,21,31,32 In our study, when the symptoms of mild and moderate bleeding were considered, the most frequently observed ones were epistaxis (33.7%), easy bruising (28.5%), and bleeding in the oral cavity (23.8%).
Central nervous system bleeding (19%) was found to be more frequent among our patients. Ragni et al 33 had reported CNS bleeding in 12 out of 75 FVII-deficiency cases. Peyvandi et al 31 had reported the frequency of CNS bleeding as 17%, whereas Lapecorella and the IRF7SG trial 32 had reported this frequency as 7%. Since our institution is a maternity and children’s hospital, we suppose that CNS bleeding episodes were detected early by the help of close monitoring in the neonatal intensive care unit.
Another severe hemorrhage type in our study was joint bleeding with a frequency of 16.6%. Its frequency had been reported as 21% to 22% in the literature.31,32 The frequency of GI bleeding episodes was found to be 7.1% in our group. Peyvandi et al 31 had reported this frequency as 14%, whereas Lapecorella and IRF7SG trial 32 had reported it as 14% and Herrmann et al 16 had found it as 17% for homozygous cases.
The lower frequency of menorrhagia among our cases might be related with the younger age of our female patients and the relatively low number of females in the study.
Out of 73 patients, only 8 cases had genetic analysis and 5 of them were homozygous, 1 was compound heterozygous and 2 were heterozygous. It is known that same genetic mutations might present with different clinical phenotypes. While one of our patients homozygous for T359M mutation presented with severe phenotype, the other with the same mutation had only moderate bleeding episodes. Severe bleeding episodes were observed in 2 of our patients with homozygous G283S mutation. Cases having severe and mild phenotypes were published with this genotype.13,16 Regarding to our compound heterozygous T324M/R353Q case who presented with mild clinical symptoms, we could not find any case with the same compound heterozygous mutation in the literature. But there are studies which report many asymptomatic cases with the R353Q mutation.34,35 In the literature, there were only 2 cases with FVII deficiency having a homozygous T324M mutation, and they were clinically asymptomatic. 36 Recently, reports of case series evaluating genotype–phenotype relations are growing in number.36,37
The minimal FVII level that will provide hemostasis is not clear. Small variations at the coagulation tests do not accurately reflect the bleeding tendency by clinical means. The patients with FVII:C levels at the lower limit of normal range may still experience bleeding episodes and may require treatment.32,38 One of our patient with FVII:C level of 54% had significant bleeding during circumcision and required treatment with rFVIIa.
The hit ratio for rFVIIa in the treatment of bleeding episodes is ranging between 76% and 100%. In the study entitled “Compassionate and Emergency Use Programmes,” Mariani et al 19 had reported that rFVIIa treatment had an efficacy of 78% in CNS bleeding, 100% in intra-artricular bleeding, and 66% in other hemorrhagic events. Similarly, surgical prophylaxis was shown to have a success rate of 96%. In the STER (Seven Treatment Evaluation Registry) study that is a multicenter prospective, observational, Web-based study protocol providing the frame for a structured and detailed data collection, 41 surgical operations (24 “major” and 17 “minor”) were performed in 34 participants with rFVIIa. Only 3 cases with low-dose rFVIIa application had bleeding during orthopedic surgical interventions. Although no thrombotic event was reported in this study, an antibody production was observed in a patient with multiple dental extraction procedures. 39
Brenner et al 20 had used an Internet-based registry (haemostasis.com) to evaluate 39 hemorrhagic episodes in 30 patients and had reported success in 26 patients treated with rFVIIa. The number of children and adults involved in both studies by Mariani et al 19 and Brenner 20 appear to be equal. Our observation suggests that rFVIIa has a better efficacy in children. It might be argued that there are other factors that make the control of bleeding more difficult in adult patients.
Two of our patients received once weekly rFVIIa prophylaxis due to recurrent CNS bleeding. One of them had been receiving rFVIIa prophylaxis for 5 years, whereas the second had been receiving it for 30 months. The dose administered was 20 mcg/kg per week. There is no prospective study providing guidance for secondary prophylactic approaches. The prophylactic doses that are used in clinical practice are usually 20-40 mcg/kg/dose and may be instituted twice a day to twice a week.24 –26,28,29 Once a week therapy was preferred due to pay-back protocols of the social insurance corporation of our country and the convenience of the intravenous route. Only two cases with once a week prophylactic use were reported as letters in the literature.26,27
Between 1996 and 2003, over 700 000 clinical applications of rFVIIa with a dose of 90 mcg/kg were reported, and among these 2 disseminated intravascular coagulation and 16 thrombotic events were observed. 40 Recombinant FVIIa is used at lower doses in FVII deficiency, so the agent can be considered to be safer in clinical use. 21 In the study of Mariani et al 19 entitled “Compassionate and Emergency Use Programmes.” no thromboembolic events were reported. Three patients for whom the presence of inhibitors were reported had a history of pdFVII use. 19 In our study, thrombotic side effects of rFVIIa were not observed during the treatment of bleeding attacks and inhibitor did not occur.
Due to the high rate of consanguineous marriage in our country, it is not surprising for us to diagnose numerous FVII deficiencies. As severe bleedings usually occur during the first year of life, it is worthy to examine bleeding diathesis of this age group cautiously. Therefore, neonatologists have a vital role in the first evaluation and diagnosis of these patients.
As a conclusion, data concerning congenital FVII deficiency in children is limited or made up of just anecdotal case reports. Hence, prospective studies concerning childhood period are required.
Footnotes
Authors’ Note
All authors contributed to data acquisition and drafting the paper.
Acknowledgments
We would like to thank Prof Dr. Guglielmo Mariani and Novo Nordisk Turkish Affiliate for their help and support they have provided during the mutation analysis of our patients.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.