
Abstract
Platelets play a crucial role in the pathogenesis of atherosclerosis, thrombosis, and stroke. Aspirin used alone or in combination with other antiplatelet drugs has been shown to offer significant benefit to patients at high risk of vascular events. Resistance to the action of aspirin may decrease this benefit. Aspirin resistance has been defined by clinical and/or laboratory criteria; however, detection by laboratory methods prior to experiencing a clinical event will likely provide the greatest opportunity for intervention. Numerous laboratory methods with different cutoff points have been used to evaluate the resistance. Noncompliance with aspirin treatment has also confounded studies. A single assay is currently insufficient to establish resistance. Combinations of results to confirm compliance and platelet inhibition may identify “at-risk” individuals who truly have aspirin resistance. The most effective strategy for managing patients with aspirin resistance is unknown; however, studies are currently underway to address this issue.
Keywords
Introduction
Platelets play a very important role in the pathogenesis of acute vascular events leading to thrombosis of the coronary and cerebral arteries. Blockage of these arteries leading to regional ischemia of heart and brain tissues precipitates heart attack and stroke. In 1897, acetylsalicylic acid (ASA) was commercially produced (and named “aspirin”) by modifying salicylic acid by a German chemist, Felix Hoffman. 1 Since the publication of University of Minnesota-educated Dr Lawrence Craven’s clinical observations, 1 clinical trials have proven aspirin’s role in preventing vascular events. Therefore, aspirin has been the drug of choice for over half a century for the primary and secondary prophylaxis of thrombotic events. Despite extensive use of aspirin as an antiplatelet medication for primary or secondary prevention of vascular thrombosis, there is considerable concern about the degree of protection it offers. In this review, we explain the phenomenon of “aspirin resistance,” discuss the limitations of aspirin therapy, and suggest methods to monitor “at-risk” individuals. Ability to monitor and determine at-risk patients will provide opportunities for the clinicians to customize antiplatelet therapies.
Aspirin Guidelines for Primary and Secondary Prevention of Stroke
Results obtained from the clinical trials have led to the acceptance of aspirin therapy for primary and secondary prevention of vascular events and stroke. Aspirin has been approved by the United States Food and Drug Administration (FDA) as 1 of the 4 antiplatelet therapy agents for noncardioembolic stroke or transit ischemic attack (TIA). 2 While approved in the United States for primary prevention, aspirin is not approved in other countries. For example, aspirin is only licensed for secondary prevention in the United Kingdom.
Published guidelines concerning primary and secondary prevention of stroke have consistently recommended the use of aspirin as the first choice of antiplatelet therapy in an appropriate group of patients. In 2011, the American Heart Association/American Stroke Association (AHA/ASA) 3 recommended the use of aspirin for the primary prevention of cardiovascular events and stroke in persons with a 10-year risk of cardiovascular events of 6% to 10% (Class I; Level of Evidence A), where the benefits outweigh the risks associated with the treatment. However, the use of aspirin is not recommended for the prevention of first stroke in low-risk persons (Class III, Level of Evidence A) nor in persons with diabetes in the absence of other established CVD (Class III; Level of Evidence B). For secondary prevention of stroke in patients with noncardioembolic stroke or TIA, 2 the AHA/ASA recommended aspirin therapy (50-325 mg/d) alone (Class I; Level of Evidence A), the combination of aspirin (25 mg) and extended-release dipyridomale (200 mg) twice daily (Class I; Level of Evidence B), or clopidogrel (75 mg) alone (Class IIa; Level of Evidence B) as viable options for initial therapy. Combined use of aspirin and clopidogrel is not recommended for routine secondary prevention of stroke and TIA (Class III; Level of Evidence A) due to increased risk of hemorrhage. In cardioembolic stroke, the use of aspirin alone is recommended only when a patient is unable to take oral anticoagulants (Class I; Level of Evidence A). The American College of Cardiology Foundation/AHA 4 recommended aspirin (75-325 mg/d) for the prevention of myocardial infarction (MI) in patients with extracranial carotid and/or vertebral arteries disease (Class I, Level of Evidence: A). If the same patient presents with a history of stroke or TIA, the same guidelines recommend the use of aspirin alone (75-325 mg/d), clopidogrel alone (75 mg/d), or the combination of aspirin and extended-release dipyridomale (25 and 200 mg twice daily), respectively (Class I, Level of Evidence: B).
Clinical Trials Using Aspirin as an Antiplatelet
Numerous clinical studies have assessed the safety and efficacy of aspirin use for primary and secondary stroke prevention. Detailed discussion of past and ongoing trails is outside the scope of this review. However, the clinical studies that have used aspirin for stroke prevention are outlined in Table 1.
Clinical Trials for the Use of Aspirin for Stroke Preventiona
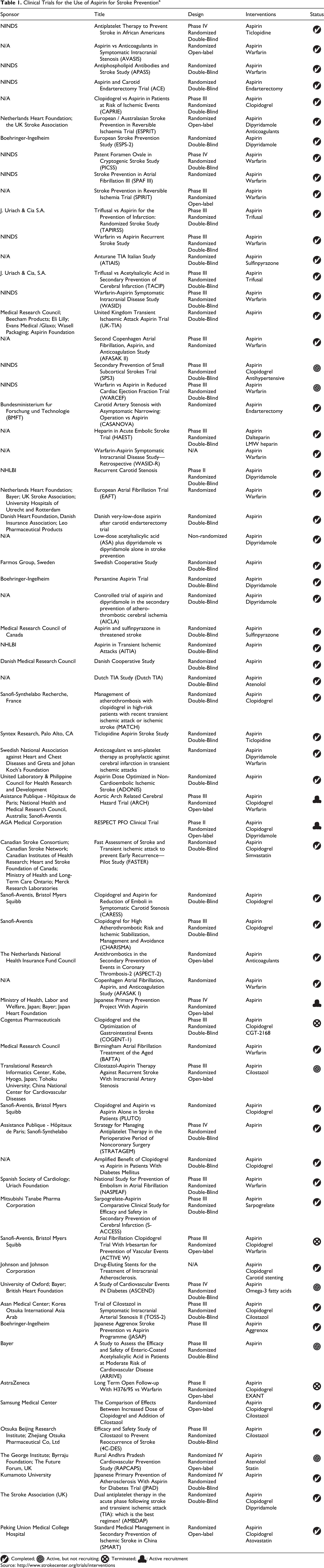
 : Completed;
: Completed;  : Active, but not recruiting;
: Active, but not recruiting;  : Terminated;
: Terminated;  : Active recruitment
: Active recruitment
Source: http://www.strokecenter.org/trials/interventions
Bleeding Complications Due to the Use of Aspirin
In recent decades, aspirin has been the most-prescribed component of antiplatelet therapy for primary or secondary prevention of MI and acute ischemic stroke (AIS). However, aspirin regimen with a therapeutic effect as an antiplatelet agent vis-à-vis its bleeding complication has been of great interest among cardiologists, stroke neurologists and primary care physicians. Inhibition of cyclooxygenase 1 (COX-1), and subsequently prostaglandin, by aspirin can result in damage to the upper gastrointestinal (GI) mucosa. 5 Several studies have demonstrated increased risk of upper GI complications at higher daily doses of aspirin. 6 In a clinical trial, 2435 patients with TIA or AIS were randomly assigned to receive 600 mg aspirin twice daily, 300 mg aspirin once daily, or a placebo. 7 The study found no difference in efficacy between 300 and 1200 mg doses of aspirin, while the lower dose led to less GI complications such as bleeding. A meta-analysis of 287 randomized trials published by Antithrombotic Trialists’ Collaboration showed that a low daily dose of aspirin (75-150 mg) was as effective as a higher dose of an antiplatelet regimen for the prevention of vascular events such as nonfatal MI, nonfatal stroke, or vascular death. 8 The same outcomes were confirmed by a systemic analysis of 31 clinical trials with a total of 192 036 patients, which suggested that the daily use of low dose aspirin (<100 mg) was associated with the lowest risk of GI and intracranial bleeding, when compared with a moderate (100-200 mg) and high (>200 mg) dose of aspirin. 6
Aspirin Resistance
Individuals taking aspirin but who fail to appropriately respond to the medication may remain at higher risk of cerebrovascular events. The term “aspirin resistance” or “aspirin nonresponse” has no standard accepted definition in the literature. The definition should be pharmacologically based on the inability of a drug to reach its target as a consequence of reduced bioavailability, in vivo inactivation, or negative interaction. 9 However, the determination has variably been based on the occurrence of a vascular event while on aspirin (clinical aspirin resistance) versus laboratory findings (laboratory aspirin resistance). 10
The lack of a clear definition has resulted in conflicting published reports on the prevalence and outcome of aspirin as an antiplatelet therapy for cardiovascular, cerebrovascular, and peripheral vascular diseases (PVD). 11 –29 There is not much data on the prevalence of aspirin resistance in generally healthy participants. In patients with various vascular diseases, the rate of nonresponders reported varies between <2% to >60%. In a systematic review of aspirin resistance studies, the mean prevalence was 24%. 30 Some studies have reported as high as 30% to 40% of nonresponders among stroke or vascular disease patients and a predicted >80% increase risk of a repeat vascular event during a 2-year follow-up period. 12 –14,20 Since the methods used to monitor aspirin resistance in these reports are not specific, the published prevalence rates are debatable. 11 –29
Clinical Aspirin Resistance
Clinical manifestation of aspirin resistance is defined as occurrence of acute vascular events such as MI, stroke, or PAD in patients despite aspirin prophylaxis. This may also be called “treatment failure.” 9,31 In the literature, it is suggested that there is an association between clinical aspirin resistance and an increased risk of vascular events. 32 –35 However, in a post hoc analysis of the data from the National Institute of Neurological Disorders and Stroke intravenous recombinant tissue plasminogen activator trial and the Trial of ORG 10172 in Acute Stroke Treatment (TOAST), aspirin treatment failure was not found to be associated with an increased risk of recurrent stroke or death. 36 The difficulty with a clinical definition is due to the implication that all acute vascular events occurring while the patient is on aspirin could have been prevented with proper response to the drug. However, aspirin inhibits only 1 pathway in platelet function and thus even an appropriate response to aspirin may not completely prevent such events from occurring.
Laboratory Aspirin Resistance
The laboratory observation of aspirin resistance definitions have ranged from the specific failure to inhibit thromboxane A2 (TXA2), failure to inhibit a test of platelet function that is dependent on TXA2 production, and the very broad failure to inhibit 1 or more platelet function assays. 9,31,37,38 The methods can predominately be divided into assays that assess the effect of aspirin on a functional platelet response and assays that measure metabolites downstream from COX-1. Studies have used single or various combinations of assays with a wide range of cutoff points for the determination of responder and nonresponder status.
For the purposes of this review, aspirin resistance is defined as the failure to inhibit either platelet function or production of a thromboxane metabolite in a laboratory study (ie, insufficient blockage of platelet reactivity despite aspirin therapy in an in vitro setting). A laboratory definition is preferred as ideally it would be desirable to be able to use testing to predict who may be at higher risk to have a vascular event while on aspirin so that measures can be taken to hopefully prevent an event that would meet the definition of clinical resistance. As the best test or combination of tests for the measurement of laboratory resistance has yet to be established, familiarity with the commonly used methods is pertinent.
Light transmittance aggregometry (LTA) measures the amount of light that passes through a platelet suspension as platelet aggregation occurs in response to an agonist. LTA is labor intensive, requires a large specimen volume, and is poorly standardized between laboratories. A variety of agonists such as arachidonic acid (AA), adenosine diphosphate (ADP), and epinephrine have been used at different concentrations and with diverse cutoff points to categorize individuals as aspirin resistant. Studies in our laboratory over 3 decades have failed to show any aspirin resistance in normal healthy participants. The only participant whose platelets failed to aggregate in response to AA stimulation was found to be deficient in platelet COX-1 enzyme activity. 39 Platelets obtained from this participant responded with aggregation when stirred with epinephrine and arachidonate, suggesting prostaglandin endoperoxides and TXA2 are not essential to cause irreversible aggregation of platelets. Aspirin resistance by LTA has been associated with an increase in adverse events. Gum et al 15 studied aspirin sensitivity by platelet response to aggregating agents such as ADP and AA among stable cardiovascular participants (n = 326) on aspirin (325 mg/day). They found that 5.5% were nonresponders to aspirin and 24% were semiresponders by platelet aggregation, in comparison 9.5% were nonresponders by Platelet Function Analyzer-100 (PFA-100; Siemens Healthcare Diagnostics, Deerfield, Illinois). In a follow-up study of these participants, 16 aspirin resistance was found to be associated with an increased risk of death, MI, or cerebrovascular accident.
Whole blood aggregometry (WBA) or multiple electrode aggregometry (MEA) uses electrical impedance to measure platelet aggregation. The use of whole blood decreases the amount of sample processing compared with LTA; however, this test also remains labor intensive and requires a large specimen volume. A study 20 conducted in Austria assessed platelet function by WBA of patients undergoing arterial angioplasty while on 100 mg aspirin per day. This study demonstrated that reocclusion at the sites of angioplasty occurred only in men for whom platelet dysfunction was evident by aggregometry.
The PFA-100 measures platelet aggregation by running whole blood under shear stress through a membrane with an opening coated with collagen/epinephrine or collagen/ADP and measuring the time to closure of the opening. The test is easy to operate; however, the PFA-100 is sensitive to multiple variables other than aspirin, including the von Willebrand factor, platelet count, and hematocrit. 40 –43 In a retrospective study of aspirin for secondary prevention of vascular events, Grundmann et al 44 observed a 34% nonresponder rate in symptomatic individuals with an event within the last 3 days. Bugnicourt et al 45 found an increased rate of early neurological deterioration in patients with AIS on 160 mg aspirin daily in nonresponders by collagen/epinephrine PFA-100 testing compared with responders.
The VerifyNow Aspirin assay (Accumetrics, San Diego, California) measures the agglutination of fibrinogen coated beads with the patient’s platelets in response to the agonist AA. The result is reported in aspirin response units. The test is an easy to use whole blood point-of-care test. Chen et al 46 found a rate of aspirin resistance rate of 27.4% in patients with stable coronary artery disease. Patients with aspirin resistance had a higher rate of a composite cardiovascular end point (15.6% vs 5.3% hazard ratio).
Plateletworks (Helena Laboratories, Beaumont, Texas) is another modification of platelet aggregation test. The test compares the platelet count in whole blood as measured by impedance in nonagonist control and agonist containing tubes. The difference in platelet count reflects the degree of aggregation and is used to calculate the percentage aggregation. The method is quick and simple use; however, there is little experience with this approach. Hurlen et al 19 used the related method of Wu and Hoak 27 to determine the platelet aggregation ratio as a marker for assessing platelet function and evaluated the effect of aspirin (160 mg/d) in 143 patients who had survived MI. Based on their definition of nonresponders to the action of aspirin, they could only identify 2 participants as primary nonresponders.
Thromboelastography (TEG; Haemoscope Corporation, Niles, Illinois) is a viscoelastic whole blood test that measures clot formation in response to thrombin generation. A further modification of this test, the TEG Platelet Mapping system (Haemoscope Corporation), assesses platelet inhibition by comparing the maximum amplitude of clot strength from a standard TEG and reactions activated with reptilase and factor XIII with and without the addition of AA or ADP. A study of patients undergoing coronary artery bypass grafting (CABG) demonstrated a 30% rate of aspirin resistance based on positive findings with at least 2 of the following tests: TEG Platelet Mapping, WBA, or 11-dehydro-thromboxane B2. 47 Nonresponders had a higher rate of graft thrombosis compared with responders (45% vs 20%, P < .05). Tantry et al 48 found a much lower rate of only 1 in 223 patients undergoing percutaneous intervention or history of stent thrombosis based on TEG Platelet Mapping and LTA.
Serum thromboxane B2 (TXB2) is a stable metabolite of thomboxane A2 (TXA2). Aspirin inhibits COX-1 activity resulting in decreased TXA2 production, which leads to lower levels of TXA2 metabolites. TXB2 has been proposed as the most specific biochemical marker for aspirin. 10,49 –51 The rate of aspirin resistance is quite low based on serum TXB2. Frelinger et al 52 observed a rate of only 2 in 682 participants treated with aspirin that presented for cardiac catheterization who had serum TXB2 levels in the range of nontreated individuals; however, it was not established if this reflected noncompliance or nonresponse. Based on receiver–operator characteristic analysis, a serum TXB2 level of >3.1 ng/mL was associated with increased risk of major adverse cardiovascular events.
Yet another test, urinary 11-dehydro thromboxane B2 (11-DTB2), may offer an advantage over the other assays. The 11-DTB2 is a stable metabolite of TXA2. 53,54 It can be measured using the FDA approved AspirinWorks test (Corgenix, Broomfield, Colorado). Testing can be performed on stored urine samples, which has logistical advantages for research studies. Urinary 11-DTB2 includes both platelet and nonplatelet sources. Monitoring stable metabolites of TXA2, such as 11-DTB2, has been performed among patients with history of vascular disease and stroke. 12,32,55 –62 Bruno et al 56 published the results of urinary 11-DTB2 of 87 African American participants within 4 months of a noncardioembolic ischemic stroke. The study suggested a significant independent effect of aspirin on urinary 11-DTB2 levels regardless of the aspirin dose (between 325 and 1300 mg daily). In a follow-up study conducted by the same group, 55 the investigators concluded that the fluctuations in urinary 11-DTB2 level were not correlated with changes in aspirin dose. However, in both the studies, underpowered sample size had been mentioned as among the shortcomings of the studies. Eikelboom et al 12 analyzed baseline urinary 11-DTB2 in 5529 patients enrolled in the Heart Outcomes Prevention Evaluation (HOPE) study. Of these patients, 488 were on aspirin regimen. On the basis of their findings, they concluded that in aspirin-treated patients, increased levels of urinary 11-DTB2 predicted future risk of MI or cardiovascular death. The patients with the highest levels of urinary 11-DTB2 had 3- to 5-fold higher risk of cardiovascular death compared with those in the lowest quartile.
The detection of aspirin resistance is greatly influenced by the method and cutoff point. Several studies have used multiple methods to look for aspirin resistance and have found large differences between methods. The prevalence of aspirin resistance is also dependent on the dose 63,64 and timing 65 of aspirin therapy. Lordkipanidze et al 66 compared 6 methods for detecting aspirin resistance in 201 patients with stable coronary artery disease. They observed poor correlation between assays and a wide range in prevalence according to the assay: 4% LTA with AA, 10.3% to 51.7% LTA with ADP, 6.75% with VerifyNow Aspirin, 18.0% withWBA, and 59.5% with PFA-100. Grove et al 50 used 6 methods (LTA with AA, MEA, PFA-100, VerifyNow Aspirin, urinary 11-DTB2, and serum TXB2). The study found poor correlation between the methods, no correlation between urinary 11-DTB2 and the 4 platelet function tests, and serum TXB2 correlated only with VerifyNow Aspirin. Six participants were nonresponders by LTA, only 2 of which were nonresponders by another method, PFA-100. Sane et al 24 evaluated the effect of aspirin (325 mg/d per month) in patients suffering from congestive heart failure by multiple methods. A participant was considered a non-responder when 4 of the following 5 parameters were observed: collagen-induced aggregation >70%, ADP induced aggregation >60%, whole blood aggregation >18 ohms, expression of active glycoprotein (GP) IIb/IIIa>220 log mean fluorescence intensity units, and P-selectin positivity >8%. Using this complex rating, persistent platelet activation was observed in 50 of the 88 patients (56.8%). However, when participants were grouped into aspirin nonresponders and aspirin responders based on this system, only 3 of these variables, ADP induced aggregation, expression of active GP IIb/IIIa, and P-selectin positivity, were statistically different among the 2 groups. A number of other variables were tested in the 2 groups by PFA-100 testing and flow cytometry with a significant difference found only with GP IIb/IIIa activity, GP Ib, platelet-endothelia cell tetra span antigen (CD151), and platelet leukocyte aggregates (CD151 and CD14).
The timing of testing must also be considered when evaluating for aspirin resistance. Henry et al 65 observed that once daily dosing of aspirin did not prevent platelet aggregation for a full 24-hour significant platelet aggregation appeared before the next dose of aspirin. Zimmerman et al 29 identified aspirin nonresponders as those who had >90% inhibition of TXA2 formation in the presence of 100 μmol/L aspirin and 1 mmol/L arachidonate in the supernatant from platelet aggregation. In patients who had undergone CABG, AA- and collagen-stimulated formation of TXB2 was the same before and after CABG, indicating oral aspirin did not significantly inhibit platelet COX-1. However, the in vitro studies with 100 μmol/L aspirin on blood obtained from these participants showed decreased TXB2 (>10%) in most samples studied. They concluded that platelet COX-1 inhibition by aspirin is compromised for several days after CABG, probably due to an impaired interaction between aspirin and platelet COX-1. This observation indicates how complex the issues are when evaluating the effect of antiplatelet drugs during and after interventional procedures.
These observations remind us of the inadequacy of the existing methods to detect what truly represents “aspirin resistance.” Moreover, studies from our laboratory have demonstrated that platelets exposed to aspirin will respond with aggregation when stirred with prostaglandin endoperoxides or thromboxane (the bioactive metabolites of AA). 67 –70 Therefore, as long as these bioactive molecules are available in the circulation regardless of their source, there exists a certain risk of developing acute vascular events leading to MI or stroke. Because of the differences in methodologies used to monitor this phenomenon and the lack of a specific assay to determine true aspirin resistance, there is considerable confusion and the true significance of this observation remains obscure. 15,18,19,22,26 –28,71 –79 It also raises the question of how we have missed this phenomenon of drug resistance for so long. Large numbers of clinical trials have demonstrated the beneficial effects of aspirin therapy regardless of the disease state. It is hard to imagine that these earlier trials missed aspirin nonresponders. On the other hand, it is quite possible that only responders to the action of aspirin received the therapeutic benefit.
Aspirin Therapy Compliance for the Prevention of Stroke
While compliance with antiplatelet therapy for prevention of stroke is essential, patient adherence to any long-term therapy, including aspirin, has been always a challenge for treating physicians. This is particularly an issue among the elderly population with a history of stroke or MI facing memory impairment or depression, as they have the highest noncompliance rate observed within the first year of the therapy. 80 –85 Upper GI complications due to the use of aspirin can be another underlying cause of noncompliance. 86 Therefore, lack of compliance can be a significant contributing factor to the failure of aspirin therapy that on many occasions are mistakenly labeled as an “aspirin resistance” or “aspirin nonresponder” case. Cuisset et al 87 found that 19 (14%) of 136 patients at 1 month after undergoing coronary stenting were nonresponders. Following observed aspirin ingestion, only 1 of the 19 patients was still found to be nonresponsive. Schwartz et al 88 and Cotter et al 89 observed similar findings. Studies that have not considered noncompliance factor may have overestimated the rate of aspirin resistance. TXB2 can potentially be used as a marker of compliance 10,49 –51 ; however, even with testing for TXB2 levels, some individuals are classified as aspirin resistant. Although challenging, this warrants the need for establishing a reliable assessment method for gauging true proportion of noncompliant patients.
Discussion
There are several contributing factors to aspirin resistance including aspirin dose, disease severity, genetic factors, inflammation, diabetes mellitus, non-COX-1–mediated TXA2 synthesis, hyperlipidemia, smoking, and interacting drugs, especially nonsteroidal anti-inflammatory drugs (NSAIDs). 90,91 There have also been some reports revealing decreased inhibition of platelet aggregation by aspirin in the female population. 92 In view of these observations, it is necessary to develop a multicenter study to look at the prevalence of at-risk population in patients undergoing antiplatelet therapy. It is important to monitor in vitro platelet function to evaluate the state of global hemostasis (combine activation of platelets and coagulation pathways) balance, in patients predisposed to hemorrhagic conditions. The decision to start antiplatelet therapy should be custom tailored based on individualized patient characteristics such as medical history and risk profile to increase the benefit of prevention strategy vis-à-vis serious adverse effects. In many cases of failure of aspirin therapy, it is not clear whether the underlying cause is nonresponse or noncompliance to aspirin therapy. However, a large portion of these cases are bundled into the nonrespondent group without proper investigation, 88,93 leading to a strong possibility that many of these cases may not be due to an inadequate platelet response to aspirin. 94 Furthermore, in many published studies, nonresponders are grouped together based on platelet function assessments.
Platelet function tests are not specific for aspirin-dependent inhibition of COX-1 and resulting TXA2 production because platelet adhesion can be mediated by other pathways (eg, ADP, thrombin, von Willebrand factor, and endothelial shear stress). 95 Several recent studies have demonstrated that despite the inhibition of platelet COX-1 enzymes, a significant number of patients on aspirin prophylaxis had increased levels of urinary 11-DTB2. 11,12 One possible explanation for this are the nonplatelet origins of TXA2 such as monocytes, macrophages, and endothelial cells within atherosclerotic plaques. 37 Regardless of the source of TXA2, what is clear from these studies is that those patients with an excess of urinary 11-DTB2 are at risk of developing acute vascular events. 12,32,60 These observations from health care providers and researchers raise a number of concerns such as our true understanding of aspirin resistance phenomena, the prevalence of aspirin resistance in the healthy population, the causes of aspirin resistance in populations with history of vascular disease, the alternative long-term treatments for patients resistant to common antiplatelet therapies such as aspirin and clopidogrel and the appropriate dose escalation and availability of point-of-care tests that are specific and cost-effective. These concerns also raise the need to develop newer and more effective antiplatelet drugs.
It is worthwhile to emphasize that the multipathway mechanism of platelet activation requires multimode antiplatelet therapy in many cases. Platelet TXA2 production through COX-2 that is not inhabited by aspirin or residual TXA2 that may have a synergistic effect on collagen activated platelets is a factors that is needed to be considered. 96
In our earlier published articles, 67 –70,97 we described how epinephrine-mediated membrane modulation restores the response of COX-1 deficient platelets and those of aspirin-exposed platelets to the action of agonists such as AA, ADP, and thrombin, independent of bioactive metabolites of AA. We also demonstrated that small quantities of endoperoxides or thromboxane generated from platelets or from some other sources could also cause aggregation of aspirin-exposed platelets. The half-life of aspirin in circulation is relatively short, and once the liver metabolizes it, the circulating salicylic acid has no inhibitory effect on platelet function. In addition, the bone marrow continuously produces fresh platelets and releases them into the blood. These newly released platelets contribute significantly to the thromboxane circulating in vivo.
In the HOPE study in which over 5500 patients were enrolled, it was found that in aspirin-treated participants, increased levels of urinary 11-DTB2 predicted a future risk of MI and cardiovascular death. 11,12 The patients with the highest levels of urinary 11-DTB2 had a 3- to 5-fold higher risk of cardiovascular death than those in the lowest quartile. The observed excess of in vivo thromboxane production may be due to an insufficient dose of aspirin in these participants, a lack of compliance, an excess production of new platelets from the bone marrow, or an altered or accelerated aspirin metabolism by these individuals. However, what is evident from recent studies is that no matter the source of this thromboxane, it puts the patients at risk of developing acute vascular events. 11 –16
In light of the fact that the normal hemostasis is to a great extent modulated by the vasoactive metabolites of AA, thromboxane generated from platelets and prostacyclin (PGI2) produced by endothelial cells, the urinary metabolites of PGI2 can also be measured. 60 Therefore, it is recommended to monitor the levels of these vasoactive molecules. This need has been well demonstrated in studies regarding NSAIDs, where the investigators tested the hypotheses that adverse cardiovascular events reported among Anti-inflammatory Prevention Trial participants were associated with increased ratio of urinary 11-DTB2 to 2′,3-donor-6-keto-PGF1 (PGI) attributable to NSAID treatments. 60 Results of these studies showed that adverse cardiovascular events were significantly associated with higher urinary 11-DTB2/PGI ratio, which seemed to derive mainly from lowered PGI.
Moreover, the issue of noncompliance with antiplatelet treatment deserves special attention. 98 Compliance should be confirmed before platelet function-based assays, such as LTA, PFA-100, and VerifyNow, which are used to evaluate aspirin response. A ratio of serum TXB2 to functional assay or other downstream metabolite may potentially identify true aspirin nonresponders and individuals who may benefit from modification of their antiplatelet therapy.
When aspirin resistance is identified, we are still faced with an unclear path of effective antiplatelet therapy. Many studies have compared the effect of aspirin versus clopidogrel or combination of both in the prevention of primary or secondary vascular events. 99 –102 A number of antiplatelet options such as clopidogrel, dipyridamole plus aspirin, and cilostazolthat are used if the patient does not respond to aspirin. Newer antiplatelet drugs such as the P2Y12 inhibitors ticagrelor and prasugrel have been investigated versus clopidogrel for the reduction of vascular events with positive outcomes. 103,104 Several studies are now in progress that are addressing the role of switching from aspirin to another antiplatelet therapy. The Aspirin Non-responsiveness and Clopidogrel Endpoint Trial (ASCET) is an ongoing study of the superiority of switching to clopidogrel versus continuing aspirin in aspirin-resistant patients with angiographically proven coronary artery disease. 105 The Research Evaluation to Study Individuals Who Show Thromboxane or P2Y(12) Receptor Resistance (RESISTOR) trial will be evaluating whether alternating antiplatelet regimens could prevent myonecrosis after endovascular coronary procedures in patients with aspirin and clopidogrel resistance. 106
In conclusion, platelet activation and the activation of the coagulation cascade is modulated by a variety of mechanisms. Due to the complexity of the atherothrombotic process, it cannot necessarily be inhibited by an antagonistic approach of 1 pathway only. Therefore, the broad definition of “treatment failure,” without considering improper/inadequate inhibition of an enzyme or receptor, must be reconsidered. 10 Thus, there is a great need to develop assays that monitor global hemostasis (combined activation of platelet and coagulation pathways). Until such a point-of-care method is available, combinations of biomarkers may best describe the hemostatic balance and better predict the risk of adverse events. A single functional platelet study is insufficient to provide this global measure due to the multiple factors unrelated to aspirin therapy that affect these results. For aspirin, the ratio of an indicator of compliance, such as serum TXB2, and a downstream stable metabolite, such as urinary 11-DTB2 and PGI2, may identify at-risk individuals who truly have aspirin resistance. 107 The downstream metabolites may provide a measure of nonplatelet sources of thromboxane that can contribute to overall hemostasis. In addition, elevation of these stable metabolites may be an indication of waxing and waning response to aspirin, perhaps due to ongoing platelet production. Our hypothesis is that a high urinary 11-DTB2 or PGI2 and low serum TXB2 will identify a population that is compliant with aspirin therapy but has persistent reactivity of the COX pathways. Further studies are essential to validate whether the levels of these metabolites will predict the risk of future acute vascular events in patients on aspirin prophylaxis. In addition, these studies are needed to develop newer and more effective alternate antiplatelet therapies for individuals who have been clearly documented with true aspirin resistance.
Footnotes
Acknowledgments
The authors thank Eliza W. Hartley for her assistance in editing the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: supported in part by the Office of the Vice President for Research and Minnesota Medical Foundation at the University of Minnesota.