
Abstract
An increasing number of mutations and polymorphisms have been identified in the von Willebrand factor (VWF) gene of patients with von Willebrand disease (VWD). Most of the sequence alterations are within exon 28, duplicated in the VWF pseudogene on chromosome 22. Genetic recombination causing the gene conversion between the VWF gene and its pseudogene is associated with multiple substitutions in the VWF gene and with VWD. In the present study, VWF gene exon 28 was analyzed in 33 patients with VWD by DNA sequencing. A total of 73% of the patients were heterozygous for p.D1472H, p.V1485L, p.1500A, p.1501F, p.L1503P, and p.S1506L single-nucleotide polymorphisms. Family analysis revealed that the gene conversion occurred between the VWF gene and its pseudogene in 3 patients. Case-control association analysis by Haploview 4.2 did not show an association between the haplotype and VWD. In conclusion, a common exon 28 haplotype in the Turkish population, which might have arisen from the gene conversion events in the founder population, was identified.
Keywords
Introduction
Congenital deficiencies of the von Willebrand factor (VWF) molecule are associated with the most common autosomal bleeding disease von Willebrand disease (VWD). 1 The VWF is a large multimeric plasma glycoprotein that is essential to maintain hemostasis in the blood. It provides the platelet aggregation at the site of vascular injury by serving as a bridge between platelets and subendothelial matrix. It also protects coagulation factor VIII (FVIII) from proteolytic cleavage in the circulation. 2 Partial deficiency of VWF is classified as type 1 VWD, complete deficiency of VWF is classified as type 3 VWD, and functional deficiency of VWF is classified as type 2 VWD. 3 The VWF is mainly expressed by endothelial cells as a 2831-amino acids long pre–pro-VWF that comprised a short signal sequence (18 amino acids), propeptide (741 amino acids), and mature VWF (2050 amino acids). The VWF is made up of repeated A, B, C, and D domains organized as D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK. 4 Different functional domains are involved in multimerization (D1 and D2 domains), in C-terminal dimerization (C2 domain), in FVIII binding (D′ and D3 domains), in collagen binding (A1 and A3 domains), and in GPIb and GPIIb-IIIa receptor binding (A1 and C2 domains). 5,6 The multimer size of VWF is regulated by ADAMTS-13 cleavage between Y1605 and M1606 within the A2 domain. 7,8
The VWF gene is located on chromosome 12 (12p13.3) and composed of 52 exons on a 178-kb genomic region. The VWF gene exon 28 is the largest exon (1.4 kb size) encoding most of the functional domains from D3 to A3. A pseudogene containing a region from exon 23 to exon 34 of the VWF gene with 97% homology is located on chromosome 22 (22q11-13). 9,10
There is a considerable variation in the levels of plasma VWF among individuals. The ABO blood group is the genetic factor affecting the VWF plasma level most with the lowest VWF level in the blood type O individuals. 11 In addition, VWF plasma level variability was linked to chromosomes 1, 2, 5, 6, and 22 in the Spanish population. 12 Ethnic origin, age, and pregnancy are the other factors affecting the VWF plasma levels. 13 –15 Genetic variability at the VWF locus accounts for 30% of the VWF plasma variance. A large number of heterogenous mutations in the VWF gene including the gene conversions with the pseudogene have been reported 16,17 and most of the mutations are detected within the exon 28 sequence. 8 Single-nucleotide polymorphisms (SNPs) in the VWF promoter region are also associated with individual variations in plasma VWF levels. 18
The VWD is a complex disease with allelic, locus, and clinic heterogeneity. Recent studies have revealed diverse molecular defects in the VWF gene associate with VWD phenotype including missense mutations, splice site mutations, deletions, and duplications. Increasing number of nonsynonymous coding polymorphisms has been observed within the VWF gene. More than 1000 SNPs have been detected throughout the gene and more than 100 of them are located within the exonic and flanking intronic sequences. 19 Multiple nucleotide substitution in the VWF gene exon 28 region due to gene conversion resulting from the recombination events between the VWF gene and the VWF pseudogene were also reported in patients with VWD. There are 2 consensus chi-like sequences (5′-GCTGGTGG-3′ and 5′-CCTGGTGG-3′) within intron 27 and exon 28 that can promote gene conversion resulting in multiple substitution within the VWF gene. 17,18
In this study of Turkish patients with VWD (including types 1, 2, and 3 VWD), exon 28 was analyzed since it is the most commonly mutated region in the VWF gene. The results demonstrated the presence of a common haplotype of exon 28 that resulted from a gene conversion event described for the first time in the Turkish population.
Methods
Ethical Approval
All procedures involving human subjects had undergone human ethic reviews and received approval by the Ege University Medical Faculty Research Ethics Commitee, Izmir, Turkey.
Study Population
Thirty-three index cases with VWD diagnosis and 13 apparently healthy blood donors were recruited in the study and informed consent was obtained from them in accordance with the Declaration of Helsinki. Laboratory tests for VWF:antigen (Ag), VWF:ristocetin cofactor (RCo), and FVIII:C were performed at Ege University Medical Faculty Children’s Hospital Hematology Laboratory Izmir, Turkey. Genomic DNA was isolated from leukocytes using PureLINK Genomic DNA Kit (Invitrogen, Grand Island, NY).
DNA Analysis
Exon 28 of the VWF gene was polymerase chain reaction (PCR) amplified in 7 overlapping regions. Primers were designed to be gene specific to avoid amplification of the pseudogene. Primer sequences and PCR conditions are available on request. DNA sequencing service was purchased from Macrogen (Seoul, Korea).
Results
The sequence of exon 28 of the VWF gene was examined in 33 patients with VWD diagnosed as types 1, 2, and 3 VWD according to the evidence-based diagnosis and management guidelines of the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). 20 The patient study population included 7, 13, 13 patients with VWD respectively. Phenotype information for the patients is given in Table 1 .
Phenotype and Observed Variations in Exon 28 of Patients With VWD.
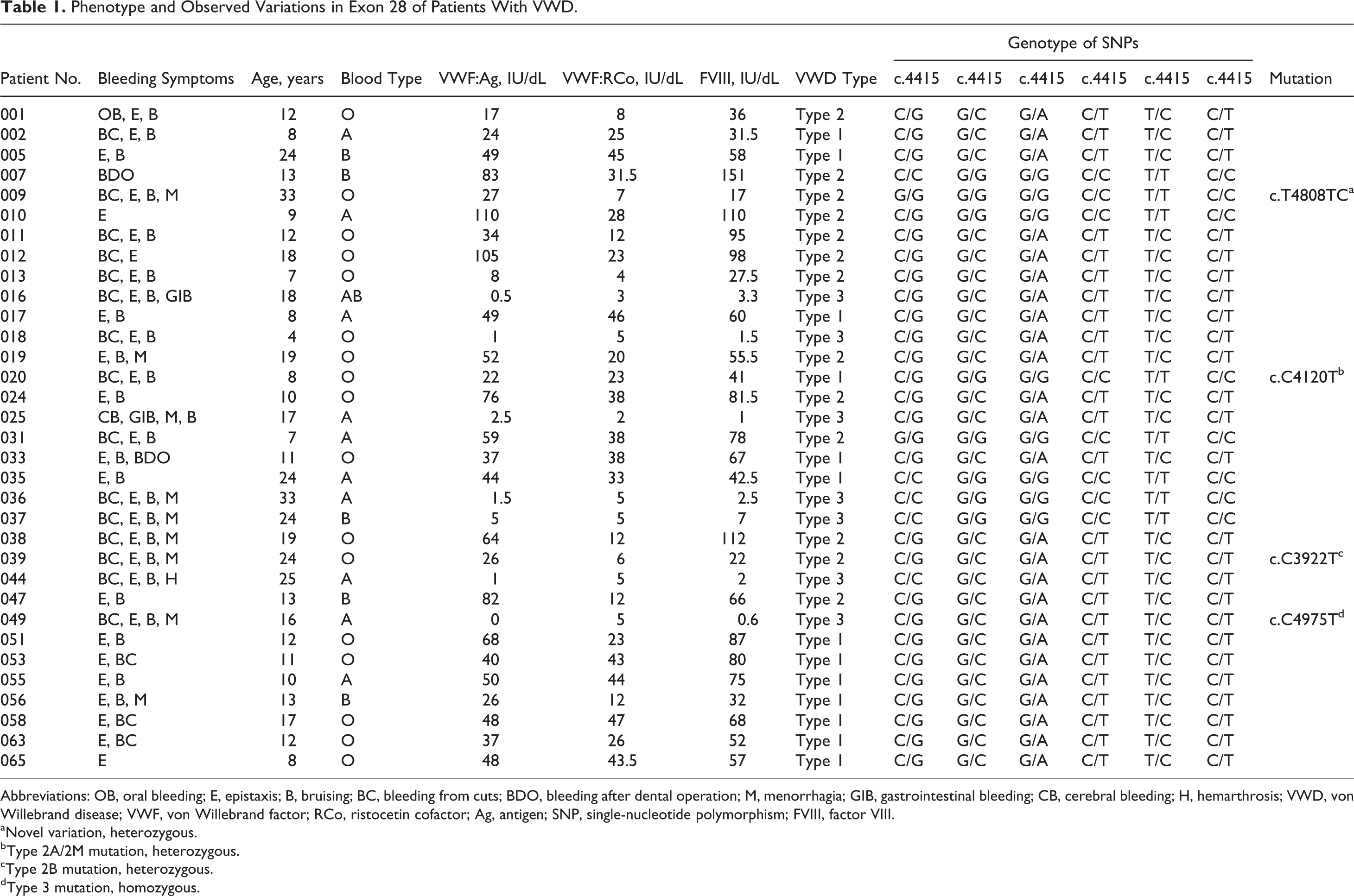
Abbreviations: OB, oral bleeding; E, epistaxis; B, bruising; BC, bleeding from cuts; BDO, bleeding after dental operation; M, menorrhagia; GIB, gastrointestinal bleeding; CB, cerebral bleeding; H, hemarthrosis; VWD, von Willebrand disease; VWF, von Willebrand factor; RCo, ristocetin cofactor; Ag, antigen; SNP, single-nucleotide polymorphism; FVIII, factor VIII.
aNovel variation, heterozygous.
bType 2A/2M mutation, heterozygous.
cType 2B mutation, heterozygous.
dType 3 mutation, homozygous.
DNA sequence analysis revealed the presence of 6 sequence changes that occurred frequently in the patient study population (Figure 1 ). These sequence and corresponding protein alterations were c.4415C>G (p.D1472H), c.4453G>C (p.V1485L), c.4500G>A (p.1500A), c.4503C>T (p.1501F), c.4508T>C (p.L1503P), and c.4517C>T (p.S1506L). Of the 33 patients, 25 were heterozygous for all the 6 alterations, 1 patient (#044) was homozygous for c.4415C>G and heterozygous for the other 5 alterations, and 2 patients were heterozygous for only c.4415G>C (#010 and #020; Table 1). c.4415G>C, c.4500G>A, and c.4503C>T sequence changes were previously known SNPs, while c.4508T>C and c.4517C>T changes were indicated as type 2A mutations in VWF mutation database (http://vwf.group.shef.ac.uk/). p.D1472H is located within the A1 domain after the C-terminal disulfide bond and all other alterations are located within the A2 domain in VWF which is involved in the ADAMTS-13 cleavage of the VWF multimers. c.4453G>C change was a novel alteration in the exon 28 of the VWF gene and it results in the substitution of valine at the position 1485 with leucine in the protein. This amino acid substitution is a conservative change since both the amino acids are neutral nonpolar amino acids. In silico analysis of c.4453G>C substitution with Exonic spling enhancers (ESE) finder (http://rulai.cshl.edu/tools/ESE2/index.html) predicted an additional binding site for the serine/arginine-rich (SR) protein SC35. However, Polyphen search revealed that it is a benign substitution (http://genetics.bwh.harvard.edu/ggi/pph/11abf76173b3e81db87de0f023350d8df887dd8b/2629832.html).
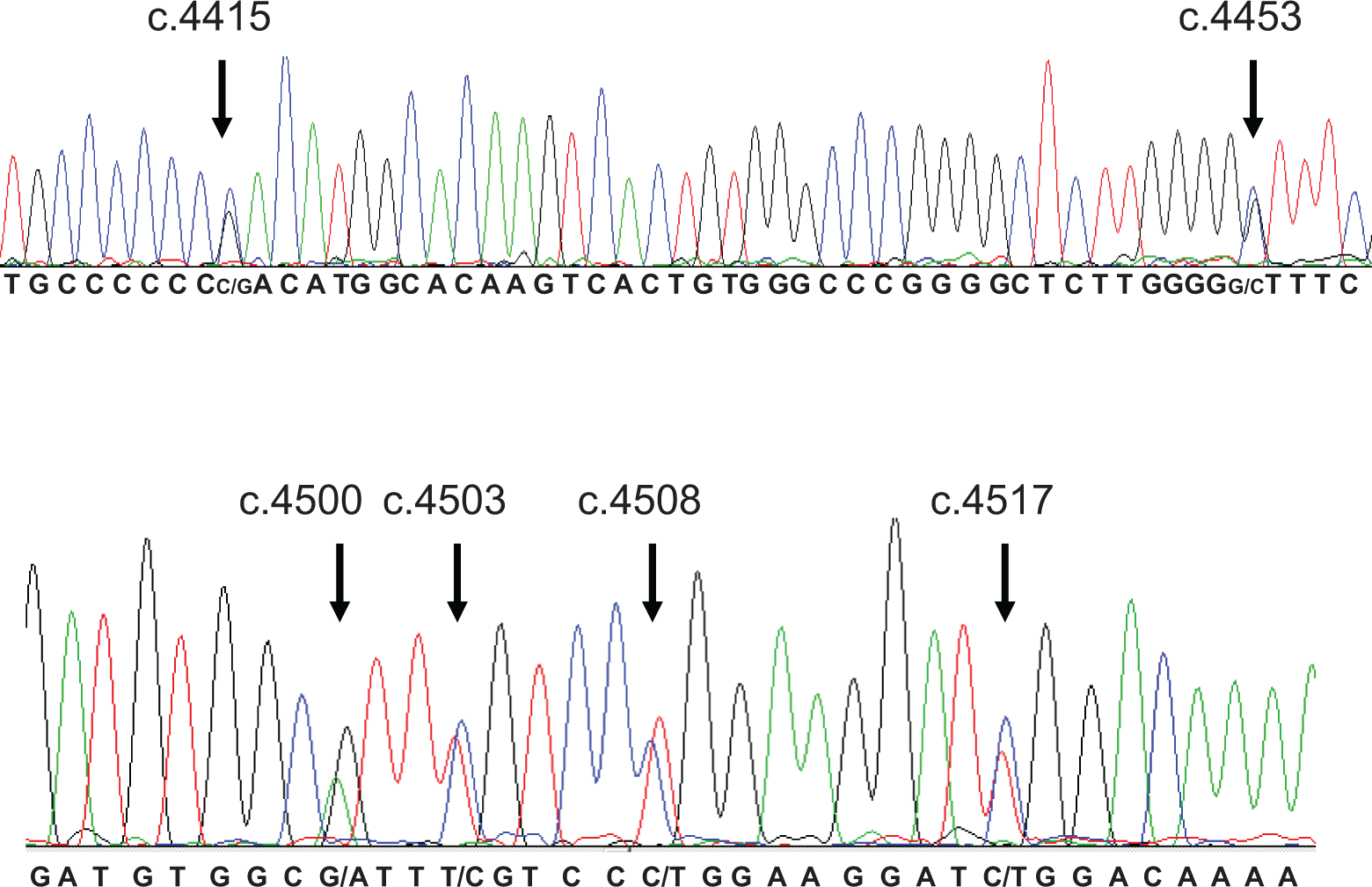
The 6 nucleotide changes in heterozygous condition in a patient with von Willebrand disease. The nucleotide changes including the novel c.4453 C/T change are shown by arrows.
None of the patients had any other VWF gene exon 28 mutations except for 4 patients: a patient with type 3 VWD (#049) was homozygous for p.R1659X type 3 mutation, a patient with type 2 VWD (#039) was heterozygous for p.R1308C type 2B mutation, a patient with type 1 VWD (#020) was heterozygous for p.R1374C type 2A/2M mutation, and a patient with type 2 VWD (#009) was heterozygous for a novel p.L1603P variation (Table 1). Levels of VWF:Ag and VWF:RCo of patients #039 and #020 are lower than the average VWF:Ag (50.93 IU/dL) and VWF:RCo (16.32 IU/dL) levels for the type 2 patients heterozygous for all the 6 sequence changes. The novel p.L1603P substitution is a conserved change, but search with the protein prediction tools polyphen, sorts intolerant from tolerant (SIFT), or AlignGVGD revealed that it is probably damaging (http://genetics.bwh.harvard.edu/ggi/pph/11abf76173b3e81db87de0f023350d8df887dd8b/2629829.html, http://agvgd.iarc.fr/agvgd_input.php, and http://sift.jcvi.org/). Additionally, in 3 patients (#035, #058, and #065), a novel polymorphism c.4483C>T (p.S1494) has been identified.
The Haploview analysis estimated 4 different haplotyopes; 5′-CGGCTC-3′ (haplotype 1, H1), 5′-GCATCT-3′ (haplotype 2, H2), 5′-GGGCTC-3′ (haplotype 3, H3), and 5′-CCATCT-3′ (haplotype 4, H4) with complete LD (Table 2 ). Both H1 and H2 are the most common haplotypes observed with 52.9% and 36.2% frequency, respectively. Family-based analysis in 12 cases revealed that 9 of the patients inherited H2 from their parents (Figure 2 ). The other 3 patients (#001, #013, and #044) had gene conversion between VWF and its pseudogene resulting in the multiple substitution in exon 28 (Figure 2). In order to determine whether H2 is associated with VWD, 13 apparently healthy individuals were screened by exon 28 sequencing. Of the 13 individuals in the normal population, 10 were heterozygous for the 6 sequence alterations. Although the size of the patient population and the normal population was small, case-control association analysis was carried out by Haploview 4.2 that calculated the chi-square statistics of SNP alleles between the 2 groups. 21 However, there was no significant association of the alleles in the patient and normal population (P > .05).
Frequency of the Estimated Haplotypes in the Study Population of von Willebrand Disease.

aEstimated frequency by Haploview 4.2.
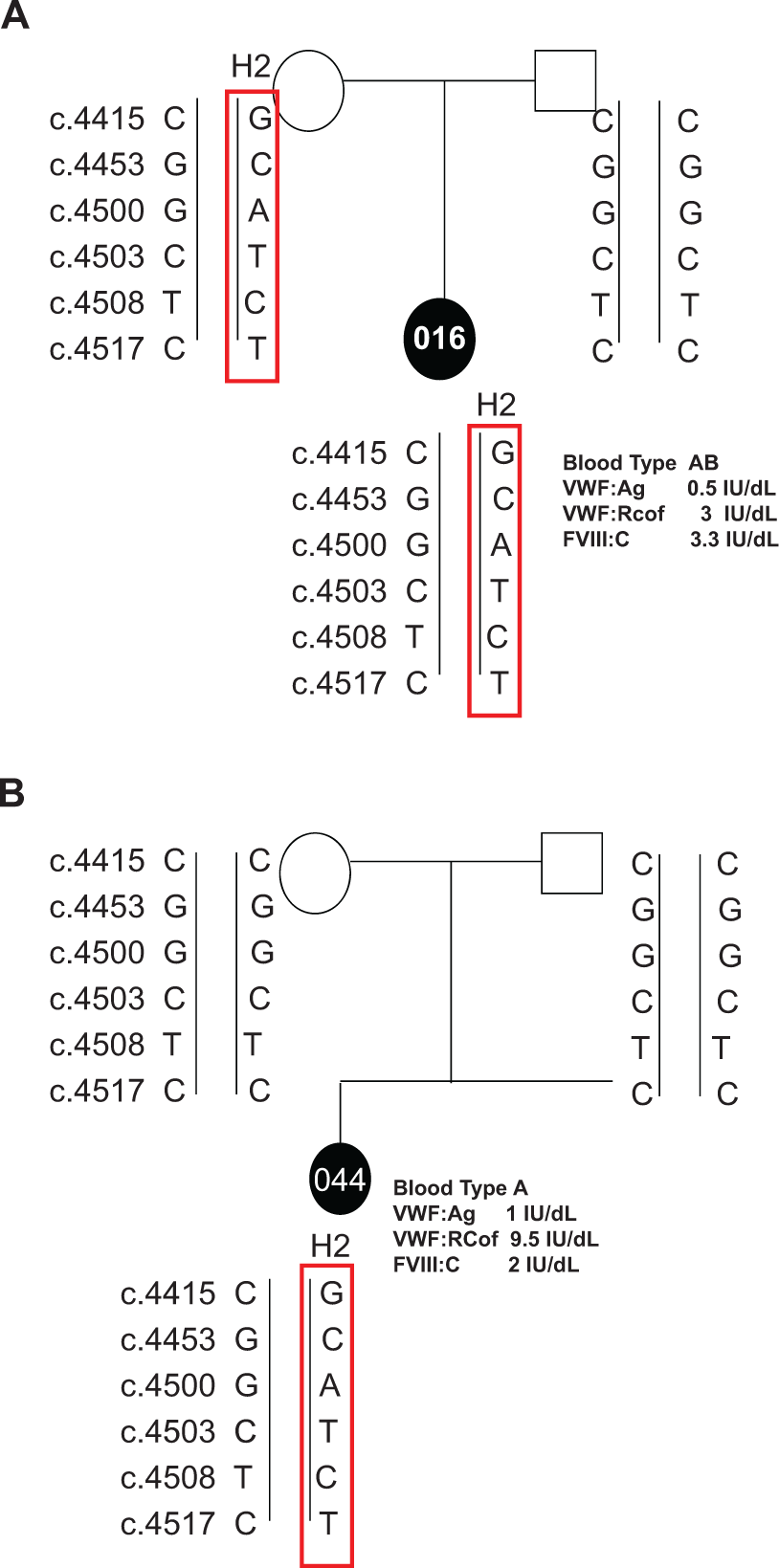
A, Inheritance of a haplotype GCATCT. B, Gene conversion between VWF and its pseudogene.
Conclusion
This study reveals a novel sequence alteration c.4453G>C (p.V1485L) in 25 patients with 5 other known variations ((c.4415C>G (p.D1472H), c.4500G>A (p.1500A), c.4503C>T (p.1501F), c.4508T>C (p.L1503P), and c.4517C>T (p.S1506L)). All 6 nucleotide changes exist in 3 common haplotypes, which were estimated by the Haploview program. Gene conversion events generating the H2 haplotype were shown in 3 families. c.4453G>C, c.4500G>A, and c.4503C>T base changes correspond to the pseudogene sequence. Gene conversion most probably started between c.4415C>G and c.4453G>C, covering a region approximately 88 bp in length.
In addition, Haploview analysis revealed the presence of a common VWF gene haplotype in the Turkish population (H2). This haplotype is observed with the same frequency in both normal and patient populations suggesting that it is a benign haplotype.
c.4453G>C is a novel SNP that causes p.V1485L substitution in VWF. p.V1485 is a nonpolar amino acid conserved between different species as well as c.4453G in the gene (human, canine, and cat). Although p.V1485L is a conservative substitution, in silico analysis demonstrated that c.4453G>C change creates an additional SR protein-binding site within exon 28. Messenger RNA (mRNA) analysis in the patients would show whether this SNP affects the splicing of VWF mRNA.
c.4808T>C is a novel variation that causes p.L1603P substitution in the A2 domain of VWF. Both c.4808T and p.1603L are conserved among different species (Pan troglodytes, Canis Lupus familiaris, Bos taurus, Rattus norvegicus, Mus musculus, Homo sapiens, Gallus gallus, and Dana rerio). Leucine and proline are neutral nonpolar amino acids; however, ring structure of proline might cause perturbation of the A2 domain folding. Besides proline substitution at this position might interfere with the ADAMTS-13 proteolysis since it is very closely located to the ADAMTS-13 cleavage site on VWF and result in the type 2A phenotype. Thus, p.L1603P substitution could be considered as a candidate mutation and in vitro ADAMTS-13 cleavage experiments would reveal whether it is associated with type 2A VWD phenotype.
p.L1503P was previously reported as type 2A mutation in a French patient who was also heterozygous for p.S1506L. Although p.S1506L was reported as a polymorphism (1%) in the French population, it was detected as a causative type 2A mutation in different populations such as American, Japanese, and Israeli. 21 Expression studies have revealed that p.S1506L increases ADAMTS-13 proteolysis of VWF in vitro. 22 However, the presence of p.S1606L with similar frequency in the patient and normal population in the present study suggest that it might not affect ADAMTS-13 proteolysis in vivo. Identification of both p.L1503P and p.S1506L changes in high frequency in both normal and the patient population in the present study suggests that they might not be responsible for the type 2A phenotype in the other populations.
p.D1472H was detected in 25 patients in heterozygous state. Two patients (#010 and #020) were heterozygous only for p.D1472H variation and 25 patients were also heterozygous for all the other 5 variations. p.D1472H is frequently observed in the African American unaffected individuals who have lower VWF:RCo/VWF:Ag ratio. However, multivariate analysis demonstrated that p.D1472H variation might affect the VWF:RCo assay without affecting the physiological function of VWF. 23 Although our patient population size is small, this study demonstrates that p.D1472H average VWF:RCo/VWF:Ag ratio was not different in the patients carrying the p.D1472H SNP (0.53) than the average VWF:RCo/VWF:Ag ratio in the noncarrier patients (0.56).
Haplotypic interactions are associated with the variable expression of the VWF gene 24 and it can be argued that the haplotype variation in association with or without pathogenic mutations might affect VWD severity as has been observed with various complex diseases such as idiopathic generalized epilepsy. 25 Previous studies have shown the presence of a founder haplotype associated with a pathogenic mutation such as p.Y1584C or p.R924Q in the patients with VWD. 26,27 Family analysis revealed that the patients inherited the 6 SNPs as a haplotype block from their families. Analysis of the 6 SNPs with Haploview estimated 3 haplotypes of which 5′-CGGCTC-3′ and 5′-GCATCT-3′ was the most common in the study population (Table 2). The low levels of VWF:Ag and VWF:RCO and FVIII:C in the patients heterozygous for these 2 haplotypes suggested that it might have an influence on the VWD phenotype. Thus, in order to determine whether the predicted exon 28 haplotype (5′-GCATCT-3′) is associated with VWD, 26 healthy chromosomes were screened for the presence of the haplotype. Haploview comparison of the haplotype frequency revealed similar estimated haplotype frequency in both normal and patient population (36.2% and 38.3%, respectively). However, analysis of the rest of the VWF gene in the patients would reveal whether this haplotype in association with a causative mutation would affect the VWD phenotype.
In conclusion, this study revealed the presence of a common haplotype in the Turkish population, containing a novel SNP (p.V1485L) and the recurrent type 2A mutations (p.L1503P and p.S1506L), which could have been produced due to a gene conversion event. In addition, although VWF gene exon 28 has been demonstrated to be the most commonly mutated region in VWD, exon 28 mutations were detected in only 12% of the patients. In addition to the recurrent type 3 p.R1659X, type 2B p.R1308C, and type 2A/2M p.R1373C mutations, a novel p.L1603P type 2A candidate mutation was detected in the Turkish patients with VWD.
Footnotes
Authors’ Note
EB: contributed to the manuscript by designing the research study, performing the research, analyzing the data, and writing the manuscript; FP: contributed to the manuscript by performing the research; MA: contributed to the manuscript by providing the patient blood samples; OYC: contributed to the manuscript by analyzing the data with Haploview 4.2; KK: contributed to the manuscript by providing the patient blood samples and revising the manuscript critically; SHC: contributed to the manuscript by designing the research study and revising the manuscript critically.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: supported by Boğaziçi University Research Fund (project no: 08HB105), Ege Hemophilia Society, and ELIPS Ltd.