
Abstract
Venous thromboembolism (VTE) is a major public health concern since the incidence of VTE rises substantially with age. Furthermore, the diagnosis can be elusive since patients can present differently, causing delay in diagnosis and initiation of treatment and resulting in major morbidity and mortality. In addition to accuracy and precision in diagnosis, antithrombotic therapies are the cornerstones of VTE management. In traditional paradigm, vitamin K antagonists (warfarin), indirect factor Xa inhibitors, and heparin are the foundation in management of VTE. Warfarin has been the only available oral anticoagulant therapy for several decades. Although warfarin is effective in both treatment and prophylaxis against VTE, there are several limitations. Therefore, the novel anticoagulation therapies, including rivaroxaban, apixaban, and dabigatran etexilate, have apparent advantages over warfarin in terms of clinical efficacy and adverse effects. The objective of this review is to describe the background and clinical implications of these novel anticoagulants.
Keywords
Introduction
Venous thromboembolism (VTE) is a major public health concern since the incidence of VTE rises substantially with age, approximately 500 cases per 100 000 persons at the age of 80 years. 1 Furthermore, the diagnosis can be elusive since patients can present differently, causing delay in diagnosis and initiation of treatment and resulting in major morbidity and mortality. In addition to accuracy and precision in diagnosis, antithrombotic therapies are the cornerstones of VTE management. In traditional paradigm, vitamin K antagonists, indirect factor Xa (FXa) inhibitors, and both unfractionated heparin and low-molecular-weight heparin, are the foundation in VTE management. The vitamin K antagonist, warfarin, has been the only available oral anticoagulant therapy for several decades. Although warfarin is effective in both treatment and prophylaxis for VTE, there are major constraints, including the need for frequent dose adjustment, a narrow therapeutic range, and significant interactions with diet and other medications.
At present, there are several new compounds being considered for VTE prevention and treatment. Rivaroxaban, apixaban, and dabigatran etexilate are the new oral anticoagulants that will be described in details in this article.
Targets of Novel Anticoagulants
The coagulation cascade consists of intrinsic and extrinsic pathways that lead to clot formation and wound closure. Factor Xa is considered a common pathway and serves as a core component in coagulation cascades by promoting thrombus formation. Therefore, inhibitors of FXa hinder thrombin generation, which will lead to diminishing the conversion of fibrinogen to fibrin. Overall, FXa is the main site of action for novel oral anticoagulants including rivaroxaban and apixaban. Conversely, dabigatran etexillate directly inhibits thrombin that prevents the conversion of fibrinogen to fibrin. Both FXa and thrombin are the major regulatory sites in coagulation cascade, which are the main sites of action of these novel oral anticoagulants.
Rivaroxaban
The Discovery
Naturally occurring FXa inhibitors were initially discovered in 1990s, which showed promising data as an alternative approach to anticoagulation therapy.2,3 In the early dawn of drug developmental process, there was no oral synthetic compounds of FXa inhibitors with adequate antithrombotic properties and good oral bioavailability identified. 4 More than 200 000 compounds were screened and eventually rivaroxaban was identified in 2008, which demonstrated significant antithrombotic activity in both in vivo and in vitro and good oral absorption in animal models. 5
Pharmacokinetics
Previous studies in healthy volunteers reveal that pharmacokinetic effects in the plasma concentration of rivaroxaban were dose dependent. The rapid absorption allows it to reach maximum inhibition of FXa activity within 1 to 4 hours after oral administration. The inhibition of FXa activity was found to range from 20% to 61% for the 5 to 80 mg doses. 6 The standard dose of rivaroxaban (10 mg) has an oral bioavailability of 80% to 100%. 7 It has a negligible interaction with diet. Thus, food restrictions are not necessary. In addition, rivaroxaban has no significant drug accumulation after its steady state. The area under the plasma concentration–time curve (AUC) and C (max) increase slightly after the administration of 40 mg rivaroxaban compared with 30 mg, but no further accumulation after administration of 50 mg rivaroxaban. 8
In terms of drug distribution, plasma protein binding is very high, almost 95% in vitro. Rivaroxaban is not expected to be dialyzable since it has very high plasma protein binding. The human plasma-to-blood partition coefficient is 1.40. The volume distribution at steady state is approximately 50 L, resulting in low tissue affinity. 9 Rivaroxaban is mainly metabolized by cytochrome P450 (CYP450) isoforms, particularly CYP3A4. Therefore, several drug interactions are important to recognize, including ketonazole and ritonavir, which are major inhibitors of CYP3A4. Rivaroxaban should be used cautiously if these potent CYP3A4 inhibitors are concomitantly used. 4 Rivaroxaban has a dual mode of elimination. Two third is metabolized by the liver to inactive metabolites and one third is excreted in urine by kidneys in unchanged forms. 4 These elimination processes result in an average half-life of 7 to 11 hours. 8
There are special considerations in some population groups. In patients with mild, moderate, and severe renal impairment, AUC increases by 44%, 52%, and 64%, respectively. Meanwhile, in patients with mild hepatic impairment (Child Pugh A), the AUC increased by 15%, and in patients with moderate hepatic impairment (Child Pugh B) it increased by 127%. In summary, caution should be taken for patients with any level of renal impairment and/or liver impairment (above Child Pugh B). 9
Pharmacodynamics
Rivaroxaban is a competitive and a direct inhibitor of serine protease coagulation FXa. Factor Xa is activated by both the extrinsic and intrinsic coagulation cascades, which exerts a crucial role in the coagulation pathway. Factor Xa converts prothrombin (factor II) to thrombin, and, finally, this complex process results in the formation of fibrin clot and activation of platelets by thrombin. The pharmacodynamic investigations demonstrated the evidence of a dose-dependent inhibition of FXa generated by rivaroxaban. The correlation between concentration and effect was well pronounced for the prothrombin time (PT), followed by the activated partial thromboplastin time (aPTT). Thus, PT may be used as a marker for anticoagulation effect of rivaroxaban. However, the association between PT and activity of rivaroxaban is poor. The relationship between PT and bleeding complications has been investigated in phase III studies which revealed the PT threshold was not accurately predicted bleeding complications in a use of rivaroxaban. In addition, rivaroxaban has no effect on thrombin content in plasma and did not interact with antithrombin. 10
Interaction studies of pharmacodynamics have been performed with rivaroxaban and different antihemostatic agents (eg, enoxaparin, clopidogrel, and warfarin). Coadministration of enoxaparin with rivaroxaban resulted in an additive FXa inhibition, as measured by anti-FXa assay. The effect of bleeding time prolongation during the coadministration of clopidogrel seems to be more pronounced. The use of low doses of rivaroxaban and warfarin confirmed that no overadditive effects were observed at the doses investigated. 10
In the case of an overdose of rivaroxaban, recombinant activated factor VII (rFVIIa) and prothrombin complex concentrate (PCC) were used in rabbit model to reverse the anticoagulant effect. 11 This study showed rFVIIa and PCC decreased aPTT and clotting time, however, these agents did not affect rivaroxaban-induced bleeding. On the contrary, a randomized controlled trial in healthy volunteers demonstrated that a single bolus (50 IU/kg) of PCC effectively normalized the PT after the administration of 40 mg rivaroxaban daily. 12
Clinical Development
Venous thromboembolism prophylaxis after total hip or knee arthroplasty. Rivaroxaban is currently approved for the prevention of VTE after elective hip and knee arthroplasty. Result from phase II trials that assessed approximately 2900 patients on different strengths of rivaroxaban revealed that rivaroxaban 10 mg once daily had the optimum balance between efficacy and safety, compared to the standard therapy of 40 mg enoxaparin once daily. 13 Rivaroxaban was further investigated in a series of 4 phase III trials that accumulatively recruited more than 12 500 patients. The studies of patients who underwent knee replacement were done in RECORD 1 14 and RECORD 2. 15 These studies demonstrated that 10 mg of rivaroxaban was superior to a once-daily dose of 40 mg enoxaparin for the prevention of VTE and all-cause mortality. Meanwhile, the RECORD 3 16 and RECORD 4 17 trials were conducted in patients who underwent hip replacement. These trials showed that 10 mg of rivaroxaban was superior to enoxaparin in both 40 mg daily and 30 mg twice daily for the same outcomes. In all 4 trials, there were no clinically significant differences in the rates of major bleeding or liver enzyme dysfunctions between the 2 regimens. Overall, rivaroxaban showed a good safety profile, including major bleeding risk and hepatic dysfunction. In addition, there is no need for a routine coagulation monitoring and dose adjustment for any demographic variables. This is consistent with the preliminary results from pooled subgroup analysis. 18 Currently, rivaroxaban was approved in the United States, Europe, and Canada for the prevention of venous thrombosis in hip and knee arthroplasty. 19
Treatment of venous thromboembolism. The efficacy and safety of rivaroxaban in the treatment of VTE, as a replacement of warfarin, were assessed in 2 phase II studies, ODIXa-DVT 20 and EINSTEIN DVT. 21 These studies suggested that rivaroxaban had similar properties in both efficacy and safety profiles compared with standard regimen. EINSTEIN DVT is a double-blind, randomized trial compared oral rivaroxaban 20 mg once daily with standard treatment of deep venous thrombosis (DVT) with subcutaneous (sc) enoxaparin followed by a vitamin K antagonist over a year of follow-up period. The first VTE events occurred in 2.1% of rivaroxaban arm, compared with 3.0% of those in enoxaparin–vitamin K antagonist arm. Meanwhile, EINSTEIN EXT studies compared 20 mg daily of rivaroxaban with placebo for an additional 6 or 12 months in patients who had completed 6 to 12 months of standard treatment of VTE. In rivaroxaban arm, the recurrent rate of VTE was less than placebo arm (1.3% vs 7.1%, hazard ratio [HR], 0.18; P < .0001). Although there was a report of the occurrence of nonfatal major bleeding in 0.7% of rivaroxaban-treated arm, there was no statistically significant difference from placebo group (P = .11). 22
Stroke prevention in patients with nonvalvular atrial fibrillation. This phase III randomized controlled trial study 23 enrolled over 14 000 patients with nonvalvular atrial fibrillation. Rivaroxaban 20 mg once daily was compared with warfarin. In the primary end points which were stroke or systemic embolism, rivaroxaban was not inferior to dose-adjusted warfarin with the rate of stroke or systemic embolism at 1.7% per year in rivaroxaban group and 2.2% per year in warfarin group (HR of 0.79, P < .001 for noninferiority). In addition, the rates of major and nonmajor clinical bleeding were comparable in both arms (14.9% per year and 14.5% per year, respectively). Despite strong clinical evidence from phase III trial supporting the usage of rivaroxaban for stroke prevention in patient with atrial fibrillation, the US Food and Drug Administration (FDA) provides recommendation against approval for this indication.
Secondary prevention in acute coronary syndrome. The ATLAS ACS-TIMI 46 trial is a randomized, double-blind phase II trial. This study assessed the safety and efficacy in almost 3500 patients who had recent acute coronary events including unstable angina, non-ST elevation myocardial infarction (MI), and ST elevation MI. The primary efficacy end point was death, MI, stroke, or severe recurrent ischemia requiring revascularisation during 6 months. The primary safety end point was clinically significant bleeding. In terms of efficacy, rivaroxaban reduced the main secondary end point of death, and subsequent acute coronary syndrome (ACS) events, however, the risk of clinically significant bleeding also increased in a dose-related manner. 24 The phase III study of low-dose rivaroxaban as adjunctive therapy in patients with ACS is currently ongoing.
Safety in Pregnancy
Rivaroxaban has shown to increase postimplantation loss and abortion in animal models. In addition, it causes several congenital malformations such as cardiac and skeletal malformations. Based on this rationale, pregnancy and breastfeeding during rivaroxaban administration are contraindicated. 25
Summary
Overall, rivaroxaban is a chemically synthetic drug that competitively and selectively inhibits the active site of FXa. Rivaroxaban has high oral bioavailability and predictable pharmacokinetic which allows convenient once-daily drug administration. There are many clinical trials in various clinical settings that support the use of rivaroxaban including stroke prevention in patients with nonvalvular atrial fibrillation, VTE prevention and treatment. However, the only clinical indication that has been approved by Europe, Canada, and US FDA is the VTE prophylaxis in hip and knee arthroplasty.
Apixaban
Background
Apixaban is an oral direct, FXa inhibitor that selectively and reversibly inhibits FXa with high-affinity binding. This agent is a potent inhibitor of free and cell-bound FXa and activated prothrombinase. 26
Pharmacokinetics
In animal models, apixaban was well absorbed in dogs, chimpanzees, and rats (mean oral bioavailability was 88%, 51%, and 34%, respectively). In human, oral bioavailability is approximately 50%. 27 Apixaban reaches peak concentration approximately 3 hours after the oral administration, while half-life is around 12 hours. It is predominantly eliminated through metabolic pathways and nonrenal mechanism. Less than 3% of the dose was excreted in bile collected between 3 and 8 hours postadministration. 28 These data suggested that biliary excretion was a minor elimination pathway. Approximately 25% (24.5%-28.8%) of apixaban was excreted in urine, which paralleled the disappearance of apixaban in plasma, which indicated that renal excretion was one of the significant routes of apixaban elimination.28,29
Apixaban is metabolized via the CYP3A4 system. Thus, coadministration of potent inhibitors of this enzyme, including macrolide antibiotics, protease inhibitors, and azole antifungal agents, should be avoided. 30
Clinical Development
Currently, apixaban has been evaluated for various indications for arthrosclerosis and thromboembolic diseases, including prevention and treatment of VTE, prevention of stroke in patients with atrial fibrillation, and prevention of cardiovascular events in ACS.
Venous thromboembolism prophylaxis
The first completed phase II trial on the thromboprophylaxis after knee arthroplasty was the APROPOS trial. 31 In this multicenter, double-blinded study, approximately 1200 patients who had elective knee replacement were enrolled. These patients were randomized to receive either 1 of the 6 different dosages of apixaban or current guideline recommended enoxaparin/adjusted dose of warfarin. Patients received the assigned treatment for 10 to 14 days. At the end of the following period, mandatory venography was performed. The primary outcome was a composite of VTE and all-cause mortality during treatment. The results showed that patients in apixaban group had lower primary outcome rates than the other counterparts. Apixaban regimens demonstrated relative risk reductions from 21% to 69% when compared to enoxaparin and 53% to 83% when compared to warfarin, suggesting this drug has a wide therapeutic window. In addition, there is no statistically significant dose response. On the contrary, there was a significant dose effect on total bleeding rates, regardless of daily or twice-daily dosing. All 4 major bleeding events that required surgical intervention were in patients receiving 20 mg of apixaban daily. Therefore, a total daily dose of 5 mg per day (2.5 mg twice a day or 5 mg daily) has a favorable benefit and bleeding risk profile which was selected for phase III study trial.
In the phase III trial, ADVANCE-1, 32 a multicenter, double-blinded randomized trial, 3195 patients who were undergoing elective knee arthroplasty were randomly assigned to receive apixaban 2.5 mg twice a day or 30 mg enoxaparin sc every 12 hours for 10 to 14 days. After the treatment, patients were again evaluated with bilateral venography. The primary efficacy outcome was a composite of any DVT, nonfatal pulmonary embolism (PE), and all-cause mortality during treatment. The rate of primary outcome was 9.0% with apixaban as compared to 8.8% with enoxaparin (relative risk, 1.02; 95% confidence interval [CI] of 0.78-1.32). Although this study did not meet the prespecified noninferiority criteria for the primary efficacy outcomes, the potential contribution factor to this finding was the relatively low incidence of the primary efficacy outcome in enoxaparin arm, which was only 55% of the predicted rate based on APROPOS study. Major bleeding and clinically relevant nonmajor bleeding were 2.9% with apixaban and 4.3% with enoxapain (P = .03). Therefore, the authors concluded that apixaban was an effective alternative to enoxaparin with similar efficacy with potentially lower rates of clinically relevant bleeding.
Meanwhile, in ADVANCE-2, 33 a multicenter, randomized, double-blind phase III study compared apixaban 2.5 mg twice a day with enoxaparin 40 mg once daily sc in patients undergoing elective unilateral or bilateral total knee arthroplasty. Apixaban was started 12 to 24 hours after wound closure and enoxaparin 12 hours before surgery. Both drugs were continued for 10 to 14 days and again bilateral ascending venography was scheduled at the end of the follow-up period. The primary efficacy end point occurred in 15.1% of apixaban patients compared with 24.4% in those treated with enoxaparin (relative risk 0·62 [95% CI 0.51-0.74]; absolute risk reduction 9·3% [5.8-12.7]). Bleeding complications occurred in 3.5% and 4.8% of patients treated with apixaban and enoxaparin, respectively (P = .09).
In ADVANCE-3, 34 a double-blind, double-dummy, randomized control trial, 5407 patients undergoing total hip replacement were assigned to receive apixaban at a dose of 2.5 mg twice daily or enoxaparin 40 mg sc daily. The study protocol was similar to ADVANCE-2 trial. Prophylaxis was continued for 35 days after surgery, followed by bilateral venographic studies. The primary outcome was the composite of DVT and nonfatal PE, or death from any causes during the treatment period. The primary outcome occurred in 1.4% in the apixaban group and 3.9% in the enoxaparin group, with relative risk reduction of 0.36 for both noninferiority and superiority. However, there is no statistical difference in bleeding risk, 4.8% and 5.0%, respectively. Based on ADVANCE-2 and ADVANCE-3 studies, apixaban was a convenient alternative for VTE prophylaxis after total hip and knee replacement without increasing the risk of bleeding. Recently, apixaban has been approved by European Commission for use in many countries in European Union for the prevention of venous thromboembolic events in patients who had elective knee or hip replacement. 35
Venous thromboembolism treatment
The Botticelli DVT 36 dose-ranging study was the first phase II study to evaluate the safety and efficacy of apixaban in the treatment of postsymptomatic DVT. In this study, 520 consecutive patients with confirmed acute symptomatic proximal DVT were randomized to receive different doses of apixaban (5 mg twice a day, 10 mg twice a day, or 20 mg once daily) for 84 to 91 days, or the comparator low-molecular-weight heparin (LMWH) as bridging to a vitamin K antagonist. The primary efficacy outcome was the composite of symptomatic recurrent VTE and asymptomatic deterioration of bilateral compression ultrasound or perfusion lung scan. The main safety outcome was the composite of major and clinical relevant minor bleeding. The primary outcome was observed after approximately 3 months of follow-up in 4.7% (95% CI 2.8-7.5) of patients in apixaban group and 4.2% (95% CI 1.4-9.6) of patients in LMWH/vitamin K antagonist group. Clinically significant bleeding occurred in 7.3% (95% CI 4.9-10.3) and 7.9% (95% CI 3.9-14.1) of patients in the apixaban and LMWH/VKA groups, respectively. Although, the results of this study showed that all 3 dose regimens of apixaban had an efficacy and safety profile similar to that of the standard therapy, the study investigators concluded that all 3 dosing regimens for apixaban showed similar efficacy and safety to the standard of care therapy. Therefore, they recommended the lowest dosage regimen of 5 mg twice daily to be used in the follow-up phase III trials.
Currently, there are 2 phase III trials underway, investigating secondary treatment of VTE. The AMPLIFY trial is the follow-up trial of the Botticelli DVT trial which involves randomization of patients to either apixaban 10 mg twice daily for 7 days followed by 5 mg twice daily or standard treatment of enoxaparin followed by warfarin for 6 months. Meanwhile, the AMPLIFY-EXT trial is investigating apixaban at 2.5 mg or 5 mg twice a day for 12 months following usual symptomatic VTE treatment versus placebo. These trials are estimated to be completed in December 2012.
Acute coronary syndrome
APPRAISE-1 37 trial was a multicenter, double-blinded trial. This phase II study investigated the effects of apixaban versus placebo in patients with recent acute coronary syndrome. These patients were randomized to 4 different doses of apixaban (2.5 mg twice daily, 10 mg daily, 10 mg twice, and 20 mg daily) or placebo for 6 months. Primary outcomes were major or clinically relevant nonmajor bleeding and secondary outcomes included cardiovascular death, MI, severe recurrent ischemia, or recurrent stroke. Before trial completion, the 2 higher dose apixaban arms were terminated due to excess bleeding, based on the recommendations of the data monitoring committee. Results from this trial demonstrated dose-dependent increase in major or clinically relevant bleeding with apixaban compared to placebo (2.5 mg twice daily [HR 1.78; 95% CI 0.91-3.48; P = .09] and 10 mg daily [HR 2.45; 95% CI 1.31-4.61; P = .005]). On the contrary, apixaban showed a trend toward a lower rate of ischemic events as compared to placebo (2.5 mg twice daily. [HR 0.73; 95% CI 0.44-1.19; P = .21] 10 mg daily [HR 0.61; 95% CI 0.35-1.04; P = .07]). The important findings from subgroup analysis in patients receiving dual antiplatelet therapy with aspirin and clopidogrel showed the risk of bleeding was more apparent and the reduction in ischemic events was less with higher daily dose of apixaban. In addition, due to the risk–benefit assessments of ischemic events and major bleeding, the investigators have selected to use apixaban at a total daily dose of 10 mg for further investigation in a definitive phase III study (APPRAISE-2). Unfortunately, the APPRAISE-2 trial was terminated early since apixaban did not significantly reduce recurrent ischemic events in patients with an acute coronary syndrome and substantially increased the risk of major bleeding. 38
Atrial fibrillation
Currently, phase III studies evaluating apixaban in patients with nonvalvular atrial fibrillation are ongoing. The AVERROES study is a randomized double-blind controlled trial evaluating 5 mg twice daily of apixaban versus aspirin in patients with atrial fibrillation who are unsuitable to vitamin K antagonist. The ARISTOTLE trial is a randomized, double-blind, parallel-arm study, evaluating efficacy and safety of apixaban in preventing stroke and systemic embolism in patients with nonvalvular atrial fibrillation by comparing apixaban 5 mg twice daily with standard warfarin therapy.
Summary
Apixaban is another potent, reversible, and highly selective inhibitor of FXa, similar to rivaroxaban. Furthermore, pharmacokinetic of apixaban is comparable to rivaroxaban including half-life, oral bioavailability, and metabolism. However, only 25% of apixaban is excreted in urine, compared with 66% of rivaroxaban. In terms of drug development, apixaban has limited supportive information from clinical trials to obtain an approval for VTE treatment and stroke prevention in patients with atrial fibrillation. Apixaban was only approved in Europe for the prevention of VTE after elective hip or knee replacement. 35
Dabigatran Etexilate
Background
Dabigatran etexilate is an orally active prodrug of the active compound dabigatran that is similar to its predecessor ximelagatran. Dabigatran etexilate is a potent, reversible direct competitive inhibitor against factor IIa (thrombin).
Pharmacodynamics
Dabigatran is a direct thrombin inhibitor that selectively, competitively, and reversibly inhibits both fibrin-bound thrombin and free thrombin. 39 Conversely, indirect thrombin inhibitors such as unfractionated heparin and LMWH cannot inhibit fibrin-bound thrombin. Therefore, dabigatran has a unique ability to inhibit fibrin-bound thrombin which theoretically benefits in terms of inhibiting coagulation cascade because fibrin-bound thrombin can continue to trigger thrombin expansion. 40
Pharmacokinetics
Dabigatran etexilate is available in capsule form. An active compound of dabigatran etexilate is coated with tartaric acid to reduce the variability of dabigatran etexilate absorption, which markedly depends on an acid environment in the stomach. Therefore, it is recommended that the capsules should not be opened, crushed, or chewed because the bioavailability can increase up to 75%. This coated composition is in the form of tiny pallets. This form of administration allows dabigatran etexilate to be absorbed independently of the acidic environment in stomach and is not essentially disturbed by coadministration of a proton pump inhibitor. 41
After oral administration, dabigatran etexilate is quickly absorbed and completely hydrolyzed to its active metabolite, dabigatran, by nonspecific ubiquitous esterases in the gut, plasma, and liver. 42 The bioconversion begins in the gastrointestinal tract, thus the drug was absorbed in the portal vein as a combination of both prodrug and active moiety. The absolute bioavailability after oral administration of dabigatran etexilate ranges from 3% to 7%. 43 After oral administration of dabigatran, the time taken to reach the peak concentration level in the plasma is approximately 0.5 to 2 hours (average, 1.5 hours). Steady-state dabigatran concentrations are achieved within approximately 3 days in healthy volunteers and there is no unexpected accumulation of dabigatran after multiple dosing. 41
In terms of metabolism, the bioconversion of dabigatran etexilate to active moiety is completed in the liver, and approximately 20% is conjugated with glucuronic acid and excreted via the biliary system. 42 As dabigatran is approximately 35% protein bound, interactions involving protein binding are not thought to be clinically relevant. 39 Dabigatran etexilate is not metabolized by the cytochrome P450 enzymes or other oxidoreductases but is a substrate for p-glycoprotein. In patients with mild liver dysfunction, the AUC is comparable to that of healthy volunteers, and the bioconversion of the prodrug was slightly reduced. 41
Renal excretion is the major elimination route. Almost 80% of circulating active and small amounts of conjugated dabigatran is excreted in the urine. Based on the rate of metabolism and excretion, the mean terminal half-life of dabigatran after oral ingestion is approximately 8 hours after a single dose and ranges from 12 to 14 hours after multiple doses. The half-life is increased to more than 24 hours in patients with a creatinine clearance of less than 30 mL/min. 44 Thus, close monitoring of the renal function every 6 to 12 months is recommended during the long-term therapy of dabigatran in patients at risk of developing renal impairment.
Clinical Development
Prevention of VTE
The phase II Boehringer Ingelheim Study in Thrombosis (BISTRO) trials demonstrated that dabigatran etexilate was effective in the prevention of VTE following total hip replacement. 45 This phase II trial is a multicenter, open-label, dose-escalating study involving 314 patients. The primary safety outcome was major bleeding. The study concluded that dabigatran has a satisfactory antithrombotic potential with acceptable safety profile. Two doses of dabigatran etexilate (150 mg or 220 mg daily) were further evaluated in phase III trials for VTE prophylaxis after hip or knee replacement.
The RE-MODEL trial 46 was a randomized, double-blind study involving 2076 patients undergoing total knee arthroplasty who received dabigatran etexilate, 150 mg or 220 mg once daily or enoxaparin 40 mg daily sc, within 6 to 10 days postoperatively. The primary efficacy outcomes were similar in all 3 treatment arms (37.7% of the enoxaparin group, 36.4% of the dabigatran etexilate 220 mg group, and 40.5% of the dabigatran etexilate 150 mg group). The incidence of major bleeding did not differ significantly between the 3 groups (1.3% vs 1.5% and 1.3%, respectively). The investigators concluded that both the doses of dabigatran etexilate (150 mg and 220 mg) were noninferior to enoxaparin based on the prespecified noninferiority criteria and have similar safety profile as enoxaparin.
Meanwhile, dabigatran etexilate was compared with enoxaparin in VTE prophylaxis following total hip arthroplasty (RE-NOVATE I and II).47,48 These 2 studies compared dabigatran with enoxaparin administered using the European approval dose of 40 mg once daily, starting the evening before surgery. The dabigatran etexilate versus Enoxaparin in Prevention of VTE Post Total Knee Replacement (RE-MOBILIZE) trial 49 compared dabigatran with enoxaparin administered using the North American approved dose of 30 mg twice daily, starting 12 to 24 hours after surgery. The primary outcome of these 3 trials was total occurrence of VTE (venographic or symptomatic), nonfatal PE, and death from all causes during treatment.
The RE-NOVATE I trial randomly assigned 3494 patients to oral dabigatran (150 mg or 220 mg) once daily or enoxaparin (40 mg once daily sc) for 28 to 35 days (median 33 days). The efficacy of dabigatran etexilate was similar to that of enoxaparin (6.7% of the enoxaparin, 8.6% of the dabigatran 150 mg, and 6.0% of the dabigatran 220 mg). The incidence of major bleeding did not differ significantly among the 3 groups (1.6%, 1.3%, and 2.0%, respectively), with P = .44 for 220 mg and P = .60 for 150 mg, compared with enoxaparin. The RE-NOVATE-II trial involved 2055 patients who were relegated to receive oral dabigatran 220 mg once daily or enoxaparin (40 mg once daily sc). The primary efficacy outcome occurred in 7.7% of the dabigatran group versus 8.8% of the enoxaparin group, risk difference (RD) being −1.1% (95% CI −3.8-1.6%); P < .0001 for the prespecified noninferiority margin. Major bleeding occurred in 1.4% of the dabigatran group and 0.9% of the enoxaparin group (P = .40). The last study in the series, the RE-MOBILIZE trial, recruited 2615 patients who randomly received treatment with either dose of dabigatran etexilate for 12 to 15 days. The rate of VTE in dabigatran group was statistically higher compared to a similar duration of treatment with enoxaparin. The incidence of major bleeding did not differ significantly among the 3 groups (1.4%, 0.6%, and 0.6%, respectively). 49
In a pool analysis of 3 major prospective, randomized, double-blind noninferiority trials comparing the efficacy and safety of dabigatran (150 mg or 220 mg once-daily) starting postoperatively with enoxaparin sc in patients undergoing hip (RE-NOVATE) or knee arthroplasty (RE-MOBILIZE and RE-MODEL), 50 the outcomes occurred in 3.3% of the enoxaparin group versus 3.0% of the dabigatran 220 mg group and 3.8% of the 150 mg group. Major bleeding occurred in 1.4% of the enoxaparin group versus 1.4% of the dabigatran 220 mg group and 1.1% of the 150 mg group. In summary, oral dabigatran was effective as sc enoxaparin in decreasing the risk of major VTE and VTE-related mortality after hip or knee replacement with similar risk of bleeding complication.
Treatment of VTE
Dabigatran was compared to warfarin in terms of the efficacy and safety for acute symptomatic VTE in RECOVER-1 trial. 51 This study randomized 2564 patients with acute VTE to 150 mg twice daily of dabigatran etexilate or warfarin (dose-adjusted to an international normalized ratio [INR] of 2-3) for 6 months after initial parenteral anticoagulation therapy for a median of 9 days (interquartile range, 8-11). The rate of primary outcome, subsequent symptomatic VTE and any VTE-related death, was 2.4% among patients assigned to receive dabigatran as compared with 2.1% in those assigned to receive warfarin (HR with dabigatran 1.10; 95% CI 0.65-1.84; absolute RD 0.4%; 95% CI −0.8-1.5; P < .001 for noninferiority). Rates of major bleeding were 1.6% and 1.9% in the dabigatran and warfarin groups, respectively (HR with dabigatran 0.82; 95% CI 0.45-1.48; P = .38), and rates of any bleeding were 16.1% and 21.9%, respectively (HR with dabigatran 0.71; 95% CI 0.59-0.85; P < .001). The number of patients who had acute coronary syndromes, abnormal liver function tests, and the number of death was similar in the 2 groups. Adverse events leading to study drug discontinuation occurred in 9.0% of patients assigned to dabigatran and in 6.8% of patients assigned to warfarin (P < .05). These data suggest that a fixed dose of dabigatran is as effective as warfarin and has a similar safety profile in the treatment of DVT and PE.
Atrial fibrillation
The Dabigatran With or Without Concomitant Aspirin Compared With Warfarin Alone in Patients With Nonvalvular Atrial Fibrillation (PETRO) trial
52
was a phase II trial designed to determine a safe dose of dabigatran etexilate in 502 patients with a diagnosis of chronic atrial fibrillation as determined by the risk of bleeding and major clinical events. This study also monitored dabigatran plasma concentrations, aPTT,
A phase III trial of the randomized evaluation of long-term anticoagulant therapy (RE-LY)53,54 which was a multicenter, prospective, open-label, randomized trial with blind evaluation, a total of 18 113 patients with nonvalvular atrial fibrillation and at least 1 additional risk factor for stroke were enrolled from 951 centers worldwide and randomly assigned to receive fixed doses of dabigatran (110 or 150 mg twice daily) in a blind fashion or adjusted dose warfarin (goal INR 2-3) in an unblind fashion. The primary outcome is any stroke events (including hemorrhagic) or systemic embolism. Safety outcomes are bleeding, liver function abnormalities, and other adverse events. The strength of this study is well balanced number of VKA experienced and naive patients and the evaluation of 2 different dosages of dabigatran. Furthermore, the study had excellent follow-up rate since only 0.1% were lost to follow-up. Dabigatran etexilate 150 mg twice daily significantly reduced the rate of primary outcomes (warfarin, 1.69% per year; dabigatran 150 mg twice daily, 1.11% per year; relative risk [RR] 0.66; P < .001 for superiority) and cardiovascular death (RR 0.85; 95% CI 0.72-0.99). The rate of major bleeding was not statistically different (3.36% per year in warfarin group, 2.71% per year in 110 mg of dabigatran, and 3.11% per year 150 mg of dabigatran; P = .31). The rate of hemorrhagic stroke was 0.38% per year in the warfarin group, as compared with 0.12% per year in the 110-mg group of dabigatran (P < .001) and 0.10% per year in the 150-mg group of dabigatran (P < .001). In terms of concomitant aspirin use, the study showed that coadministration of aspirin and dabigatran increased the risk of major bleeding compared with dabigatran alone (HR, 1.91; P < .001) without any evidence of benefit in decreasing stroke and other vascular events. 51 However, aspirin use did not interact with treatment as increased bleeding risk with aspirin use was observed for all 3 treatment groups (both doses of dabigatran etexilate and warfarin), regardless of age or creatinine clearance. Similarly, renal impairment increased the risk of bleeding with dabigatran etexilate but again there was no treatment interaction. 55
Interestingly, the rates of MI were marginally higher in both doses of dabigatran etexilate compared with warfarin (0.53% per year in the warfarin group; 0.72% per year in the 110-mg group of dabigatran (RR 1.35; 95% CI 0.98-1.87; P = .07) and 0.74% per year in the 150-mg group (relative risk 1.38, 95% CI 1.00-1.91; P = .048). One of the possible explanation might be that warfarin provides better protective benefit against coronary ischemic events than dabigatran. 56 However, the rates of MI were similar between patients with atrial fibrillation who were receiving the early generation of direct thrombin inhibitor, ximelagatran, and those who were receiving warfarin. 57 Thus, the author concluded that the reason for the higher rate of MI was unclear.
In terms of adverse effects, dabigatran etexilate showed no evidence of liver toxicity. However, rates of dyspepsia were higher in dabigatran (11.8% with 110 mg BID and 11.3% with 150 mg BID) compared with warfarin (5.8%). Based on RE-LY trial, the benefit of 150 mg dosage of dabigatran etexilate in reducing overall stroke and 110 mg in lower bleeding risk compared with warfarin were consistent, regardless of the variation in INR control by the study centers among patients assigned to receive warfarin. 58 However, for secondary outcomes such as all vascular events, nonhemorrhagic events, and mortality, the advantages of dabigatran in RE-LY were more pronounced in patients with poor INR control than those with good INR control. In subgroup analysis in patients with prior stroke or transient ischemic attack, 150 mg of dabigatran comparatively reduced stroke or systemic embolism compared with warfarin (2.78% per year and 2.32% per year, respectively). 59
Dabigatran has been approved by FDA for stroke prevention in atrial fibrillation. However, intracerebral hemorrhage (ICH) is a major concern since there is no effective antagonist for dabigatran. However, PCC has been studied in murine model of ICH associated with dabigatran. 60 This study demonstrated strong evidence that PCC effectively prevents intracerebral hematoma expansion in a murine model. However, the role of PCC in humans was inconsistent with the animal model. Eerenberg et al randomly allocated healthy volunteer who received dabigatran 150 mg twice daily, followed by a single bolus of PCC. This study concluded that PCC has no influence on the anticoagulation action of dabigatran. 12
A cost-effective analysis of dabigatran compared with adjusted-dose warfarin for preventing ischemic stroke in elderly patients >65 years with nonvalvular atrial fibrillation, based on data from RE-LY trial and other relevant publications of anticoagulation suggested that dabigatran is more cost effective compared with warfarin. The incremental cost-effectiveness ratio compared with warfarin was $51 229 per QALY (quality-adjusted life year) for low-dose dabigatran and $45 372 per QALY for high-dose dabigatran. 61 However, there were several limitations of this study. First, event rates were mostly derived from a single randomized clinical trial and extrapolated to a 35-year time frame clinical trials, with approximately 2 years of follow-up. Second, the cost of dabigatran was estimated on the basis of pricing in the United Kingdom.
Acute coronary syndrome
A phase II dose-escalating trial for Dabigatran Etexilate in Patients With Acute Coronary Syndrome (RE-DEEM) was a double-blind, placebo-controlled study which randomized 1861 patients with acute coronary syndrome and at least 1 cardiovascular risk factor to either placebo or dabigatran at the 1 to 4 dosages (50 mg, 75 mg, 110 mg, and 150 mg) twice daily, starting within a few days (mean 7.4 days) after acute coronary events and continuing for 6 months. In this study population, 99.2% were on dual antiplatelet therapy. The primary outcome was the composite of major or clinically relevant minor bleeding during the 6-month treatment period. The study showed that combined treatment of dabigatran and dual antiplatelet therapy was associated with a dose-dependent increase in bleeding events (HR 1.77 [95% CIs (0.70, 4.50)] for 50 mg; HR 2.17 [0.88, 5.31] for 75 mg; HR 3.92 [1.72, 8.95] for 110 mg; and HR 4.27 [1.86, 9.81] for 150 mg). However, dabigatran significantly reduced coagulation activity and may have benefit to reduce cardiovascular events when added to dual antiplatelet therapy in the doses of 110 to 150 mg twice daily (3% in 110 mg and 3.5% in the 150 mg dabigatran group, and 3.8% in the placebo group. 62
Summary
Dabigatran etexilate is an orally active prodrug of the active compound dabigatran which is currently considered as a potent reversible direct inhibitor against factor IIa (thrombin). Among other novel anticoagulation therapies, dabigatran has relatively poor oral bioavailability and needs acidic environment to facilitate absorption. Renal excretion is a major route of drug elimination. In current clinical setting, dabigatran has been approved for use in preventing VTE in patients who have undergone elective total hip and knee arthroplasty in Europe, Canada, and United States. 63 In addition to VTE prophylaxis, the FDA approved dabigatran etexilate in 2010, for prevention of stroke in patients with nonvalvular atrial fibrillation 64 after the results of the RE-LY study (Tables 1 and 2).
Characteristics of New Oral Anticoagulation Therapies
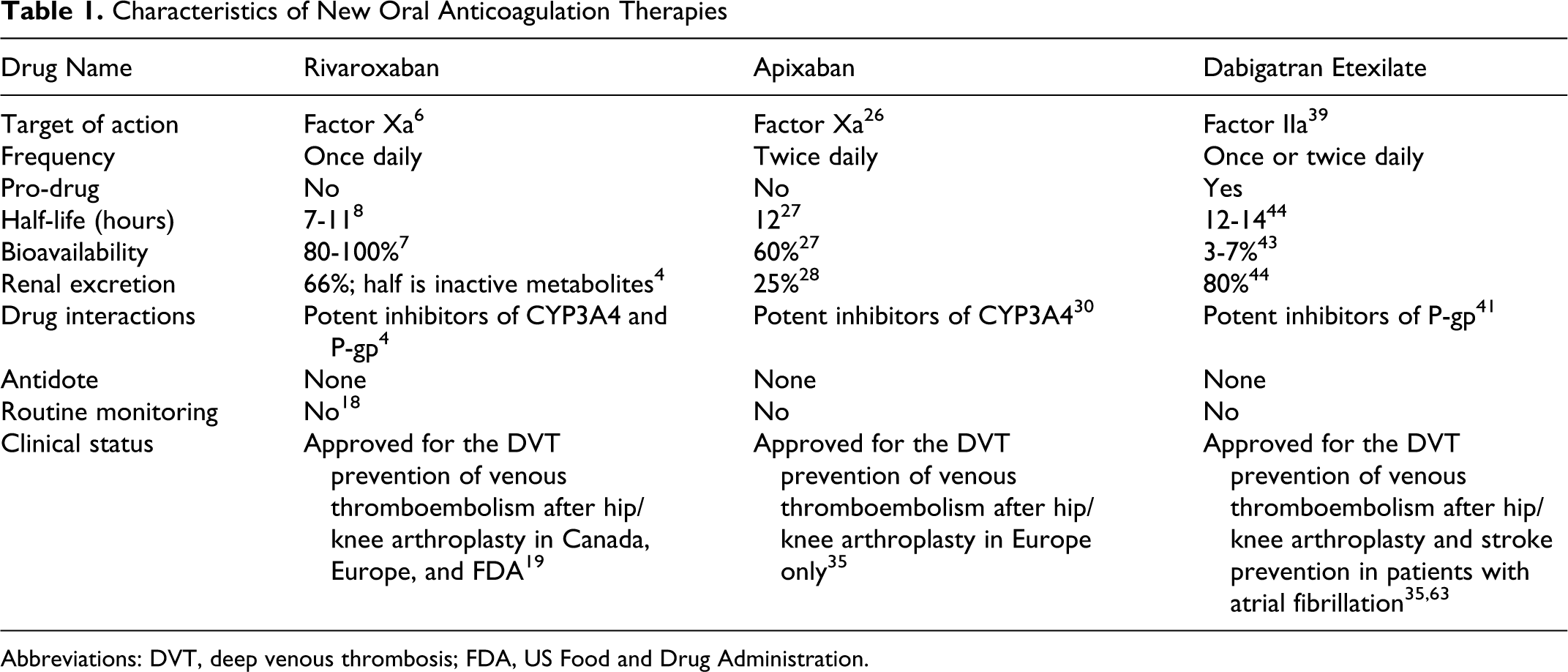
Abbreviations: DVT, deep venous thrombosis; FDA, US Food and Drug Administration.
Ongoing Phase III Randomized Controlled Trials of Novel Oral Anticoagulants (as of August 2011)
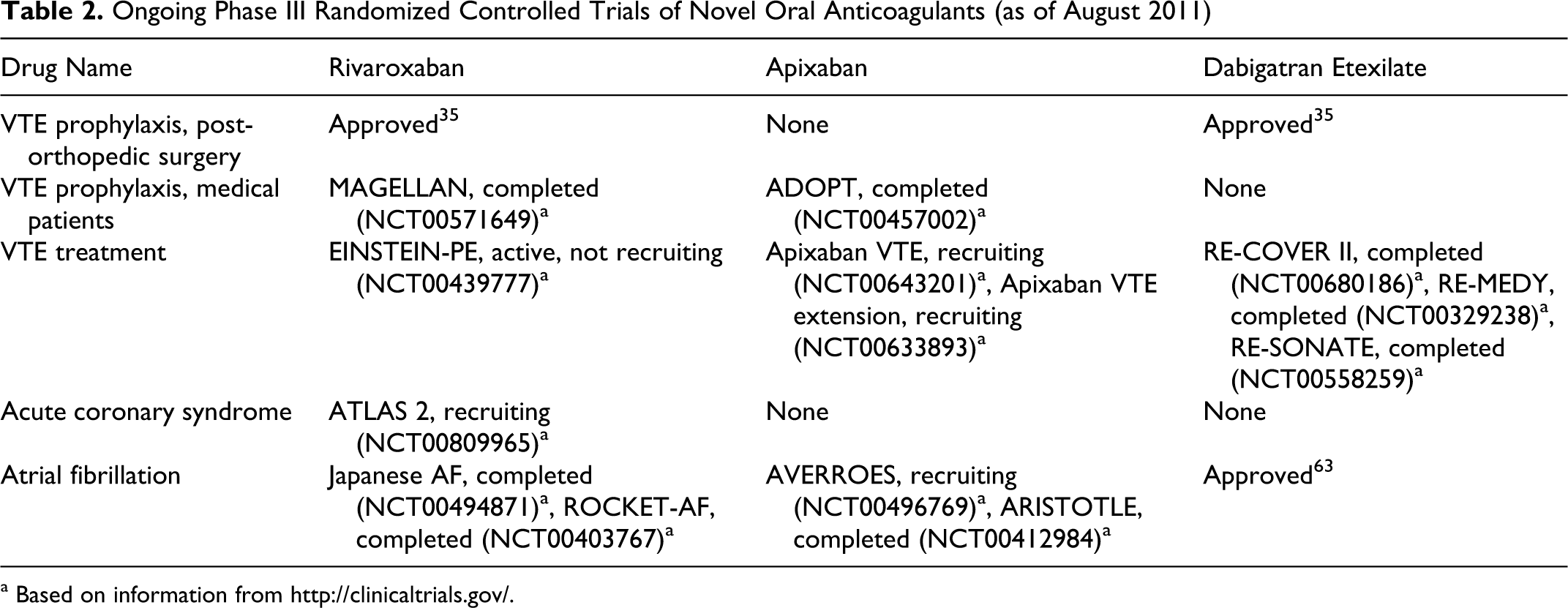
a Based on information from http://clinicaltrials.gov/.
Conclusion
For several decades, warfarin has been an exclusively available oral anticoagulation therapy. It has been proved to be an effective treatment in various clinical settings including VTE treatment and prophylaxis, stroke prevention in atrial fibrillation, and so on. However, there are several drawbacks to consider using warfarin including the need for dose monitoring and adjustment which tremendously complicates its clinical management. Until several years, novel anticoagulants were released and they could potentially address these defects associated with warfarin. There are several aspects of novel anticoagulants that need to be addressed. The upcoming studies will be needed to investigate the potential role of these medications in other clinical settings, for example, postsurgical VTE prophylaxis in abdominal surgery, prosthetic heart valves, peripheral artery disease, and cerebrovascular diseases.
In addition to rivaroxaban, apixaban, and dabigatran etexilate, other FXa inhibitors are being evaluated. Otamixaban has completed phase II trial for short-term use in percutaneous coronary prevention 64 and in non-ST elevation acute coronary syndrome. 65 The promising results are further verified in an ongoing phase III trial in patients with unstable angina or MI with non-ST elevation undergoing early invasive strategy compared with unfractionated heparin, epifibatide, or matching placebo (NCT01076764). In addition, idraparinux is a parental form of polysaccharide indirect FXa inhibitors with very long half-lives 66 that were developed for once-weekly dosing for several conditions that require long-term or chronic therapy.67,68
These novel anticoagulants seem to have better efficacy and safety against VTE and stroke prevention in various clinical settings. However, there are some major disadvantages of these novel anticoagulants. First, they have no antidote in the case of drug overdose. Second, since most of them are excreted in the urine, they can potentially cause major bleeding in patients with renal dysfunction. Therefore, using these novel anticoagulants may need careful monitoring in postmarketing surveillance studies for emerging side effects after FDA approvals.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.