
Abstract
Patients with multiple myeloma (MM) are at relatively high risk of developing thromboembolic events such deep venous thrombosis (DVT) where thalidomide therapy has been identified to increase this risk. Defibrotide (DF), a polydisperse oligonucleotide, showed previously to counteract the alterations in endothelial cells (ECs) induced by lipopolysaccharide. It prompts us to investigate the impact of thalidomide on ECs and whether DF modulates changes in fibrinolysis induced by thalidomide. In this in vitro study, MM by itself alters the profibrinolytic potential of ECs decreasing the tissue plasminogen activator (t-PA) and increasing the plasminogen activator inhibitor 1 (PAI-1) levels which is potentiated by thalidomide. Defibrotide was able to counteract these effects. Additionally, DF upregulated the t-PA and downregulated PAI-1 gene expression modulated by thalidomide. Defibrotide also protects ECs from thalidomide-mediated cell death without interfering with its antitumor effects. These findings support DF clinical use for the prevention of DVT induced by immunomodulatory drugs.
Introduction
Multiple myeloma (MM) is a progressive hematologic disease characterized by excessive numbers of abnormal plasma cells in the bone marrow and overproduction of monoclonal proteins in more than 90% of patients. 1 It accounts for 10% incidence and 20% mortality of all hematological malignancies. 2
Despite the development of several new therapies and substantially effective novel combination regimens, myeloma remains an incurable disease. Recent advances in the treatment of MM have resulted in improved response rates and overall survival in patients with MM. These advances are largely due to thalidomide-, lenalidomide-, and bortezomib-based combinations that have improved response rates not only in patients with untreated disease but also in those with relapsed and/or refractory myeloma. 3
Thalidomide was the first agent with mild immunomodulatory activity approved in 2006, by Food and Drug Administration (FDA), for use in combination with high-dose dexamethasone (DEX) in treating patients newly diagnosed with MM; single-agent therapy is commonly employed in the settings of relapsed/refractory MM. Since then, several studies have been done combining different drugs for treatment of MM; however, there are several complications related to these therapies. Patients with MM are at relatively high risk of developing thromboembolic events (TEEs), where thalidomide has been identified as a major cause of venous thromboembolsm (VTE) in patients with MM, especially when it is used in combination with other treatments such as DEX.4,5 The majority of thrombotic events described in patients receiving thalidomide have been venous, but occasional arterial thrombotic events have also been reported. 6 Thalidomide has a wide spectrum of biological effects, including some that have been hypothesized to promote thrombosis such as transient reduction in soluble thrombomodulin levels during the first month of therapy 5 and restoration of endothelial cell (EC) protease-activated receptor (PAR-1) expression after damage due to the cytotoxic agents such as doxorubicin. 7 However, the mechanisms by which thalidomide predisposes to thrombosis are not well understood and difficult to establish because of the concurrent presence of many prothrombotic factors in patients with MM. 5
Defibrotide (DF) is a purified polydisperse mixture of 90% single-stranded oligonucleotide manufactured by controlled depolymerization of DNA derived from porcine intestinal mucosa with a molecular weight distribution of 17 ± 4 kDa. Defibrotide has a number of pharmacological effects that contribute to its endothelial protective properties with profibrinolytic, antithrombotic, anti-ischemic, antiadhesive activities but no significant systemic anticoagulant effects.8–10
Defibrotide exerts minimal systemic anticoagulant effects, yet consistently appears to induce numerous antithrombotic and profibrinolytic effects both in vivo and in vitro.10–13 Preclinical studies demonstrate that DF promotes fibrinolysis via upregulation of tissue plasminogen activator (t-PA) and tissue factor pathway inhibitor (TFPI).8,10 It reduces circulating levels of plasminogen activator inhibitor 1 (PAI-1) in lipopolysaccharide (LPS)-stimulated ECs. 8 Furthermore, DF increases the plasmin activity. 14
Additionally, recent in vitro studies demonstrated that DF has no direct anti-MM activity against MM, however, in the in vitro cocultures of MM cells with bone marrow stromal cells (BMSCs), DF enhances the anti-MM activity of certain drugs, such as melphalan or DEX. This effect is associated with suppressed adhesive interaction between the 2-cell compartments; abrogation of nuclear factor-kappaB (NF-kB) activation triggered by MM-BMSC adhesion; decreased production of proliferative/antiapoptotic cytokines as vascular endothelial growth factor (VEGF) and interleukin 6 (IL-6) in the supernatant of coculture systems; and perturbed expression and/or function of key mediators of MM-microenvironment interactions, such as heparanase, angiogenic cytokines, and adhesion molecules. 15
Importantly, these findings suggest that DF is not only compatible with conventional and many novel antitumor treatment strategies but also indicate DF exhibits microenvironment-modulatory properties that serve to attenuate key aspects of tumor pathophysiology and drug resistance.
The endothelial protective properties of DF combined with its antitumor activity compatible with MM chemotherapies prompted us to hypothesize that it could counteract the modification of antithrombotic phenotype of ECs induced by thalidomide without interfering with thalidomide tumor toxicity effect.
In addition to the antitumor properties of DF, in the current study we demonstrate that DF counteracts the modification of antithrombotic phenotype of ECs induced by thalidomide.
Material and Methods
Cell Culture and Reagents
The human dermal microvascular endothelial cell line CDC/EU HMEC-1 (HMEC) was kindly provided by the Centers for Disease Control and Prevention (Atlanta, Georgia) and has been established as previously described.
16
The HMEC were cultured in MCDB 131 medium (Gibco BRL, Karlsruhe, Germany) supplemented with 10% fetal calf serum, 2 mmol/L
RPMI 8226 MM cell line was purchased from American type cell collection (ATCC, Manassas, Virginia) and cultured in RPMI 1640 modified medium (ATCC) supplemented with 10% fetal bovine serum and antibiotics.
All the cell lines were grown in humidified air containing 5% CO2 at 37°C and were subcultured through trypsin-mediated detachment every 2 to 3 days. Cells were serially passed in T25 flasks and seeded in plates for the experiments at passages 8th to 20th.
Cell Coculture System
A policoated coculture system (Becton Dickinson, New Jersey) has been used to evaluate whether MM cells by themselves can modify the antithrombotic phenotype of ECs and the effect of thalidomide (Sigma-Aldrich; 100 µg/mL) and DF (Gentium, Villa Guardia, Italy; 150 and 400 µg/mL) on this system.
The cocultures of MM cells with ECs were set up, culturing HMEC (3×105) in 6 well plates for 24 hours in the presence or absence of MM cells (3 × 106). After that, the cells were unstimulated or stimulated with 100 µg/mL of thalidomide with and without 150 µg/mL of DF.
The cell supernatants were collected for immunoenzymatic analyses (enzyme-linked immunosorbent assay [ELISA]) and the RNA were extracted for gene expression evaluation.
Endothelial Cell Fibrinolytic Parameters
To study the impact of thalidomide on ECs and whether DF modulates changes in fibrinolysis induced by thalidomide, HMEC (6 × 104) were incubated with thalidomide (50 and 100 µg/mL) in the presence and absence of 150 µg/mL of DF for 24 hours. In parallel, the coculture of ECs with MM cells was applied to evaluate the impact of MM on EC fibrinolytic properties.
Levels of t-PA and PAI-1 in the supernatant were measured by ELISA, using commercially available kits (Hyphen-Biomed-Cabru and Instrumentation Laboratory, Italy, respectively) according to the manufacturer’s instructions.
Molecular Profile Studies
Total RNA was isolated by RNeasy Mini kit (Qiagen, Hilden, Germany), according to the manufacture’s instructions, from HMEC (6 × 104) unstimulated or stimulated with 100 µg/mL of thalidomide in the presence or absence of 150 µg/mL of DF for 24 hours. The 1% agarose gel electrophoresis, stained with ethidium bromide, was performed to check the purity of the RNA.
Total RNA, 2 µg, was used for complementary DNA (cDNA) synthesis through iScript cDNA synthesis kit (Bio-Rad, California) per the manufacturer’s instructions. SYBER-green (Supermix - Bio-Rad) real-time polymerase chain reaction (PCR) was performed using an iCycler MYIQ sequence detector (Bio-Rad) in a total volume of 20 µL, according to the manufactures guidelines.
The primers pairs used were tPA forward 5′-TGACGTGGGAGTACTGTGATGTG-3′ reverse 5′-GGCGATGTCGGCGAAGA-3′; PAI-1 forward 5′-TGCCCATGATGGCTCAGA-3′ reverse 5′-GGCAGTTCCAGGATGTCGTAGT-3′ and for β-actin forward 5′-TCACCCACACTGTGCCCATCTACGA-3′ and reverse 5′- CAGCGGAACCGCTCATTGCCAATGG-3′. The expression of the genes was quantified by normalizing to β-actin expression (housekeeping gene).
Fibrin Clot Plate Assay
The fibrin clot was formed in the flat wells of microtiter plate to determine the fibrinolytic activity of cell supernatant to degrade the fibrin clots. This assay was performed according to a modified protocol. 17 Briefly, the fibrin clot was formed by mixing fibrinogen (2 mg/mL), plasminogen (37.5 µg/mL), and thrombin (1 IU/mL; Calbiochem-Nova Biochem Corp, California). This was allowed to clot for 30 minutes at room temperature. When the clot was completed the supernatant of the cells, treated with thalidomide (100 µg/mL) with and without DF (150 and 400 µg/mL), and a chromogenic substrate S-2251 (0.6 mmol/L; Chromogenix, Molndal, Sweden), were added to the clot. The microplate was incubated at room temperature for 30 minutes. Fibrinolysis takes place at the boundary of the solid fibrin matrix and the t-PA, released by the cells, penetrates the clot, and encounters plasminogen bound to fibrin. The plasmin generated by the t-PA, present on cell supernatant, was able to digest fibrin and also hydrolyze the substrate. The generation of p-nitroaniline was monitored by following the increase in OD at 450 nm in a 2.5-minute step for a period of 10 to 25 minutes in a microplate reader set at 37°C (Victor 3, Perkin Elmer, California, USA). Plasmin activity was determined from a plot of absorbance versus time as previously described. 14
Cell Proliferation Assay
Proliferation and viability of endothelium and MM cells were determined by the (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT, Sigma-Aldrich). This method is based on the cleavage of tetrazolium salt by mitochondrial dehydrogenase. For this assay, 100 µL of HMECs (or MM cells suspension) were seeded onto 96 wells plate (10 000 cells/well) and allowed to attach overnight at 37°C and 5% CO2. The culture medium was then aspirated, and fresh medium containing vehicle or thalidomide (10-25-50-75-100-500 µg/mL) with and without DF (150 µg/mL) was added to each well and left for 24 hours. Following incubation, 5 µL of MTT (5 mg/mL) was added to the wells and left for 3 to 4 hours. Plates were then centrifuged at 1200 rpm for 5 minutes and the supernatant was discarded. The dye was solubilized with 200 µL of a mixture DMSO/ethanol (1:1) and added to each well, after which the plates were vigorously shaken. The amount of MTT formazan produced during the incubation was measured by a microplate reader (Victor 3, Perkin Elmer) at 540 nm. A blank well containing only media and drug was also run as control.
Apoptosis Assay
An established method for detecting apoptosis in ECs was performed as previously described 18 using flow cytometry (FACScan and CellQuest software; Becton Dickinson, Germany). Both MM and HMEC cells were left unstimulated or stimulated with 100 µg/mL of thalidomide in the presence or absence of 150 µg/ml of DF for 24 hours. Afterward, the cells were washed in phosphate-buffered saline (PBS) and were stained with the necrosis-detecting dye propidium iodide ([PI]; 0.2 µg/mL; Sigma-Aldrich). Apoptotic cells were identified by PI-negative staining and by a characteristic side scatter (SSC) image distinct from that of nonapoptotic cells.
Statistical Analysis
The significance of differences between experimental values was assessed by means of Student t test.
Results
Defibrotide Modifies the t-PA and PAI-1 Levels in ECs Treated With Thalidomide
Enzyme-linked immunosorbent assays have been performed for determination of t-PA and PAI-1 levels in the supernatant of HMEC cells unstimulated or stimulated with thalidomide in the presence or absence of DF. In parallel, we established an in vitro model where MM cells and microvascular ECs were cultured in close proximity to each other, with the HMEC cells cultured in the lower and MM cells cultured in the upper chamber of a dual-chamber in vitro system. This system was applied in order to evaluate whether MM by themselves can alter the antithrombotic phenotype of ECs as well as the effect of thalidomide and DF on this system.
Figure 1A illustrates the results of t-PA antigen level in the supernatant of HMEC cells after 24-hour incubation in the absence (saline, control) or presence of 50 and 100 µg/mL of thalidomide. Thalidomide significantly reduced, in a dose-dependent manner, the release of t-PA in HMEC cell supernatants (P < .001). The presence of DF at 150 µg/mL (Figure 1B) modified the response of HMEC cells by significantly increasing t-PA levels reduced using both the concentrations of thalidomide (P < .01).
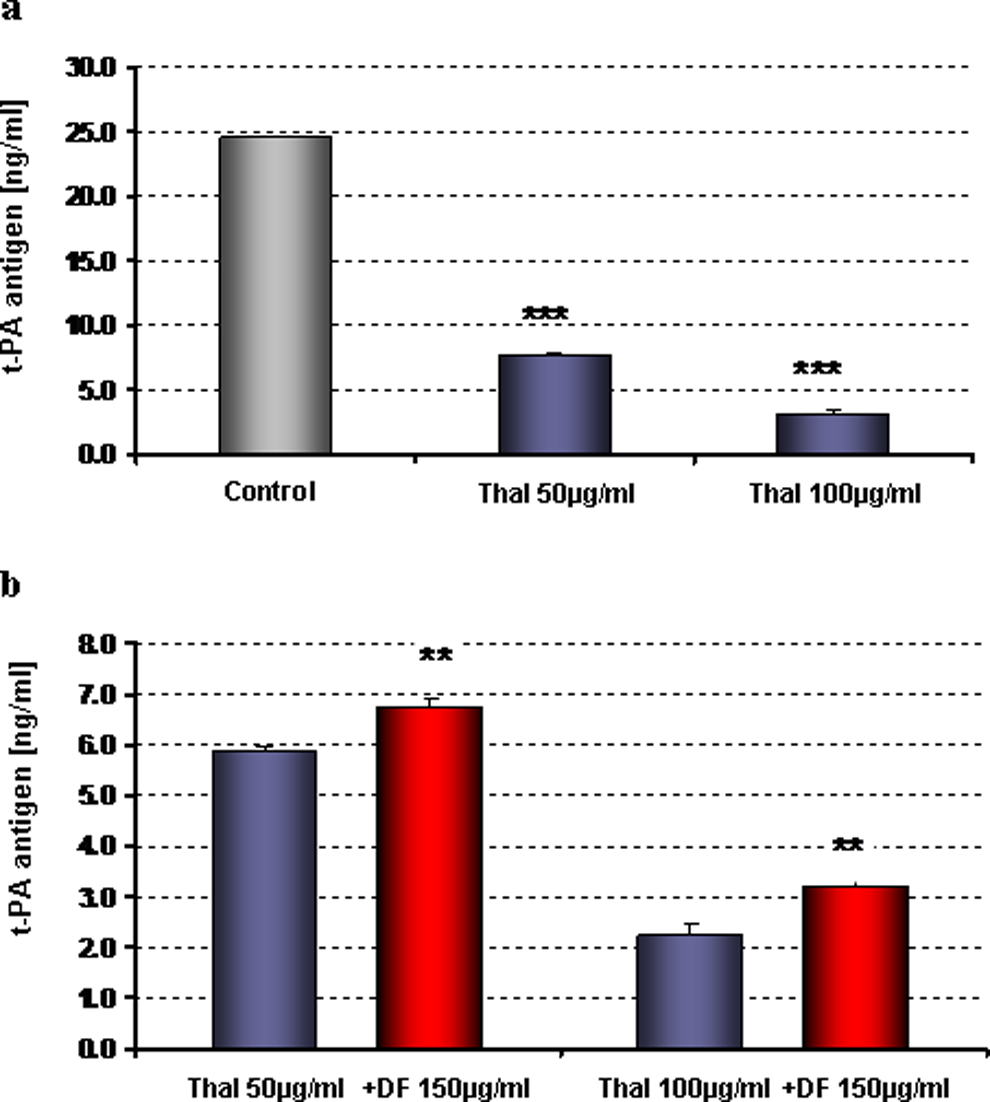
Effects of defibrotide (DF) on the t-PA release by HMECs stimulated with thalidomide. HMEC were incubated with increasing concentrations of thalidomide with and without defibrotide. A, Thalidomide decreases the release of t-PA in HMECs in a dose-dependent manner (P < .001 vs control). B, Defibrotide significantly increases the release of t-PA in HMECs treated with thalidomide (P < .01; [thalidomide + DF]-treated cells versus thalidomide alone). t-PA indicates tissue plasminogen activator; HMECs, human dermal microvascular endothelial cells.
In the coculture of HMECs and MM cells, we demonstrated that MM cells, by themselves, changes the profibrinolytic potential of ECs. In these experiments, interaction with MM triggered a significant decrease in the release of t-PA (Figure 2A) as an induction in the expression of PAI-1 in human microvascular ECs (Figure 3A). In this same system, thalidomide dramatically potentiates the MM effect decreasing the levels of t-PA (Figure 2B) and increasing the levels of PAI-1 (Figure 3B) released by HMEC cells (P < .001). Importantly, DF was able to reverse these events in activated ECs induced by thalidomide.
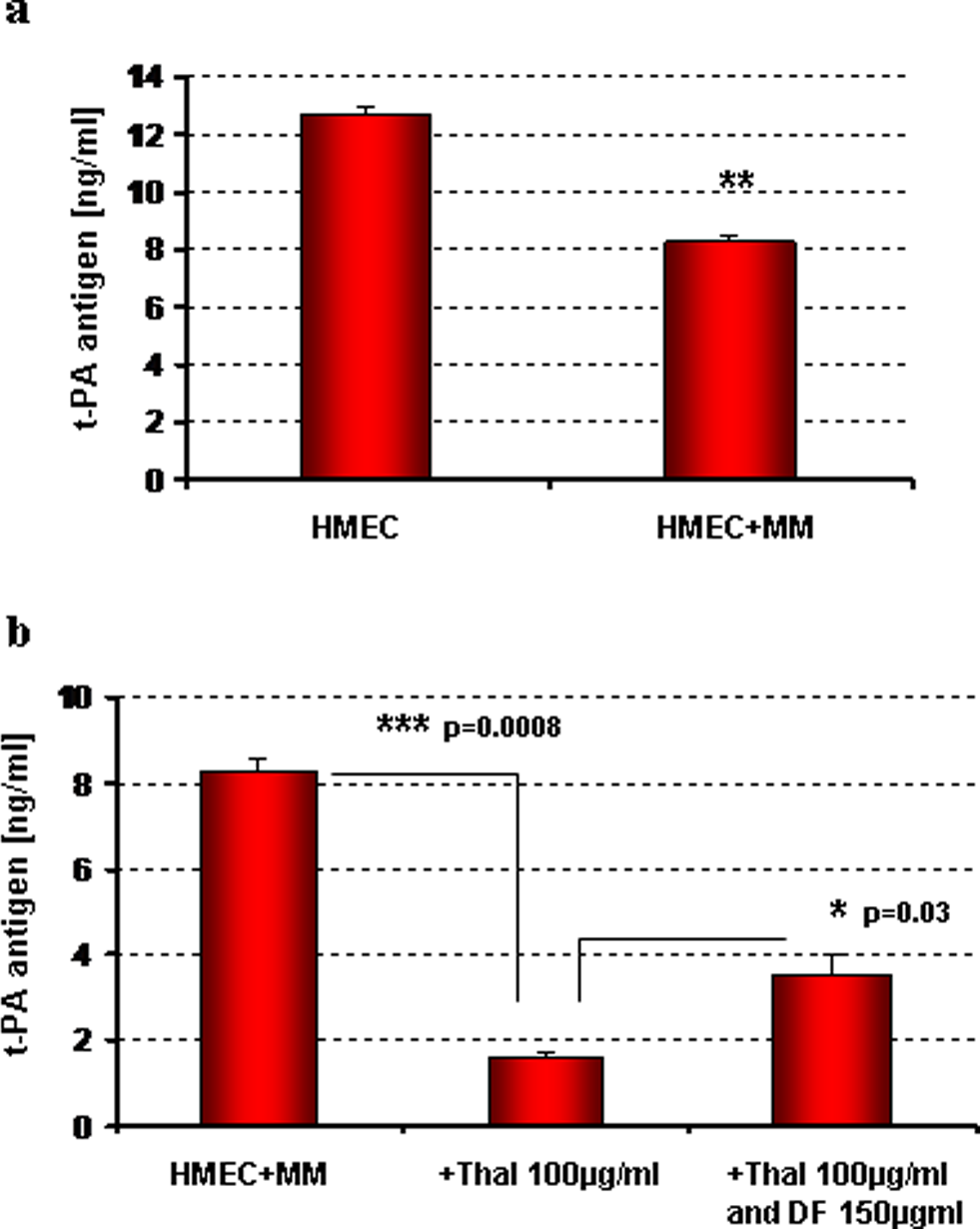
Modification on the release of t-PA by HMEC cocultured with MM cells in the presence and absence of thalidomide and defibrotide. The levels of t-PA antigen were measured by ELISA in the cell supernatants. A, MM cells change the profibrinolytic potential of ECs decreasing significantly the release of t-PA (P < .01 [HMEC + MM] vs HMEC alone). B, In that system, the thalidomide dramatically potentiates the MM effect (P = .0008); defibrotide treatment reverses this event significantly increasing the release of t-PA by thalidomide-stimulated HMECs (P < .05 [thalidomide + defibrotide]-treated cells vs thalidomide treatment alone). t-PA indicates tissue plasminogen activator; HMECs, human dermal microvascular endothelial cells; ELISA, enzyme-linked immunosorbent assay; ECs, endothelial cells; MM, multiple myeloma.
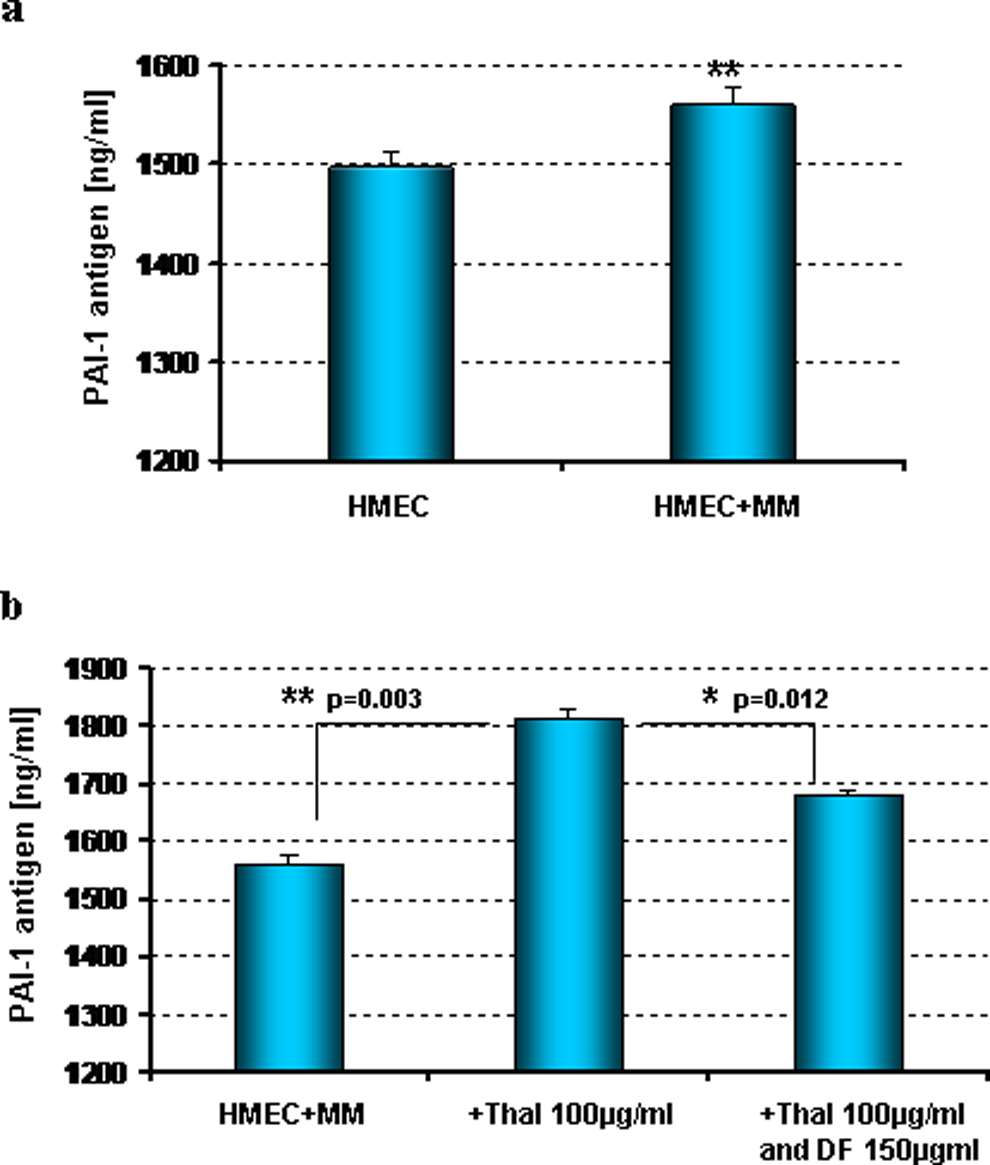
Modification on the release of PAI-1 by HMECs cocultured with MM cells in the presence and absence of thalidomide and defibrotide. The levels of PAI-1 antigen were measured by ELISA in the cell supernatants. A, The interaction with MM cells triggered in HMECs an induction in the expression of PAI-1 (P < .01 [HMEC + MM] vs HMEC alone). B, In that system, thalidomide potentiates the MM effect (P = .003); defibrotide counteracts the antifibrinolytic action of thalidomide, significantly decreasing the release of PAI-1 by HMECs induced by thalidomide (P = .012 [thalidomide + defibrotide]-treated cells vs thalidomide treatment alone). PAI-1 indicates plasminogen activator inhibitor 1; HMECs, human dermal microvascular endothelial cells; ELISA, enzyme-linked immunosorbent assay; ECs, endothelial cells; MM, multiple myeloma.
Defibrotide Upregulates t-PA and Downregulates PAI-1 Gene Expression in ECs Treated With Thalidomide
Additional studies have been performed to investigate whether DF can modulate the expression of t-PA and PAI-1 transcripts in microvascular ECs. In these experiments, the t-PA and PAI-1 messenger RNA (mRNA) expression in activated HMEC cells were quantified using real-time PCR. The HMEC cells were either unstimulated or stimulated with 100 µg/mL of thalidomide in the presence and absence of 150 µg/mL of DF for 24 hours.
As shown in Figure 4A, thalidomide reduced by 2.2-fold the t-PA expression compared with unstimulated (control) cells, whereas DF abrogate this effect upregulating by 8.8-fold the expression of t-PA reduced by thalidomide.
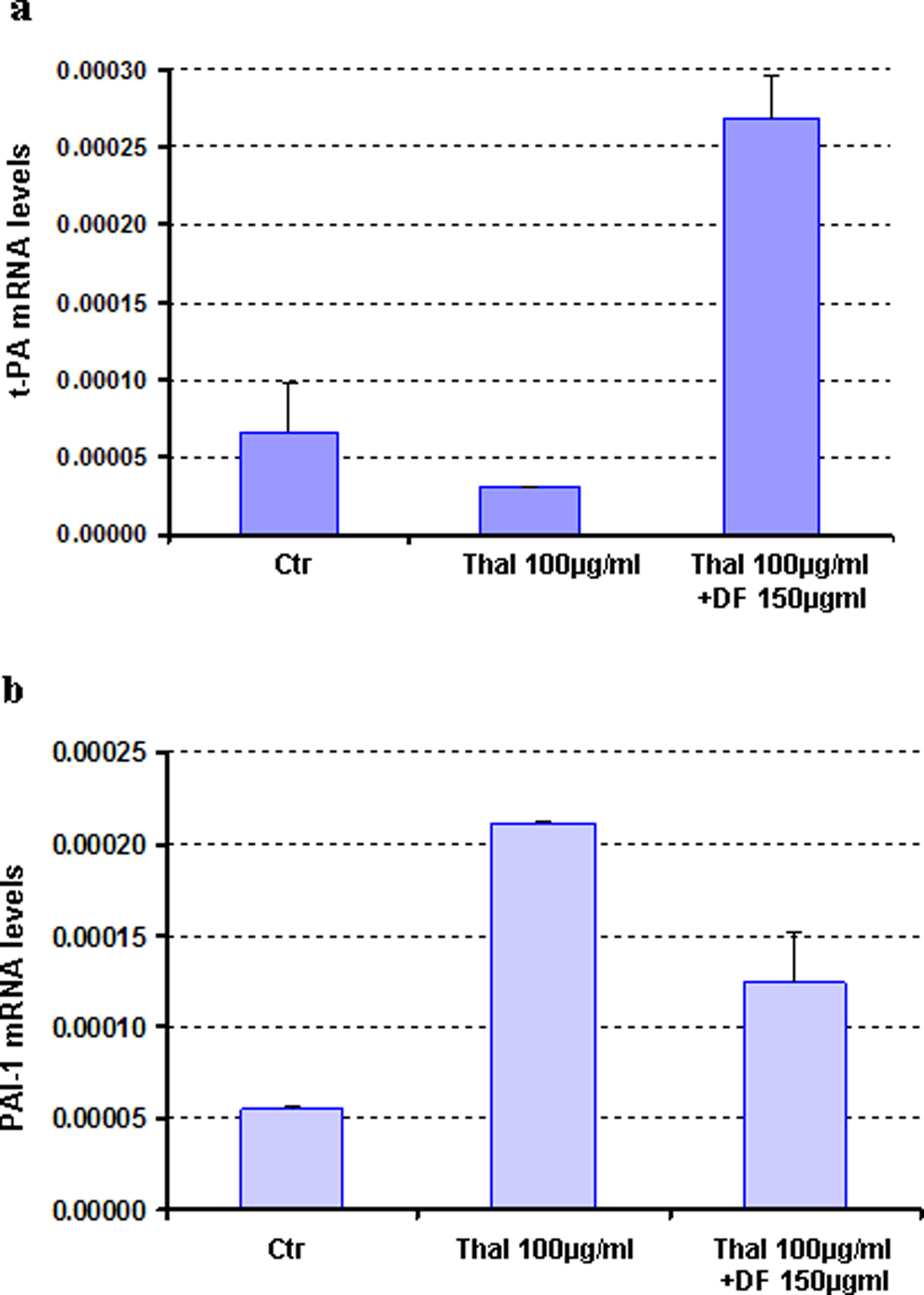
Effects of defibrotide on the t-PA and PAI-1 gene expression in endothelial cells treated with thalidomide. Total mRNA was extracted from HMECs, unstimulated or stimulated with thalidomide with and without Defibrotide for 24 hours. A, Thalidomide reduces by 2.2-fold the t-PA gene expression compared with unstimulated cells, whereas defibrotide counteracts this upregulating effect by 8.8-fold the expression of t-PA in the thalidomide-activated HMECs. B, Additionally, defibrotide is able to downregulate by 2.0-fold the PAI-1 mRNA levels in HMECs induced by thalidomide (4.0-fold of increase in PAI-1 mRNA levels). PAI-1 indicates plasminogen activator inhibitor 1; t-PA, tissue plasminogen activator; HMECs, human dermal microvascular endothelial cells; ELISA, enzyme-linked immunosorbent assay; mRNA, messenger RNA.
In addition, PAI-1 mRNA levels were upregulated 4-fold in ECs treated with thalidomide (Figure 4B). Importantly, DF reversed thalidomide effect downregulating by 2-fold the PAI-1 gene expression in the activated ECs.
Defibrotide Counteracts the Decrease in Fibrinolytic Activity of HMEC Cells Induced by Thalidomide
A fibrin clot plate assay has been performed to determine whether DF modulates the fibrinolytic activity of thalidomide-activated microvascular ECs to degrade the fibrin clot reflecting the physiological situation encountered during thrombolytic condition. The principle of this method is that in the first stage the fibrin clot is formed by mixing fibrinogen, plasminogen, and thrombin. The supernatant of the HMECs treated with thalidomide (100 µg/mL) in the presence and absence of DF (150 and 400 µg/mL) and a chromogenic substrate (S-2251) were added to the clot. The plasmin generated by the t-PA present in the cell supernatants is able to degrade the fibrin clot and also hydrolyze the substrate S-2251.
Results presented in the Figure 5 show that thalidomide reduced the fibrinolytic activity of HMEC cells and DF abrogate this effect restoring significantly (P < .01), and in a dose-dependent manner, the fibrinolytic capability of ECs to degrade the fibrin clot.
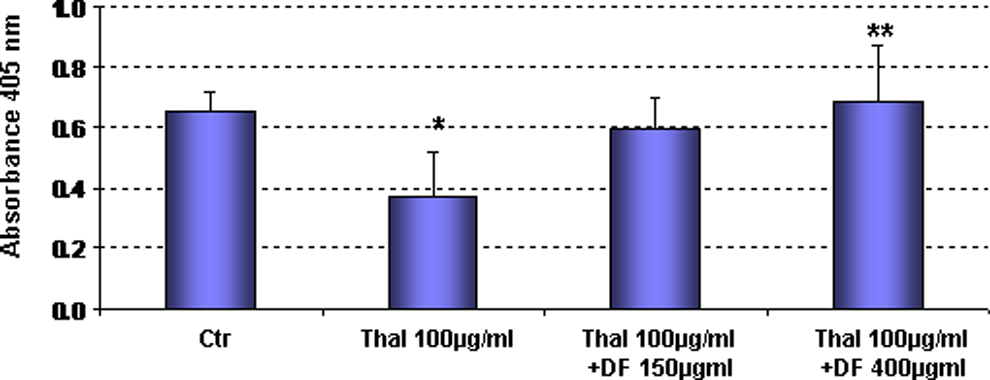
Effects of defibrotide on fibrinolytic activity of thalidomide-activated endothelial cells. HMECs were treated with thalidomide in the presence and absence of defibrotide, and a fibrin clot plate assay has been performed. Thalidomide reduces fibrinolytic activity of HMECs and defibrotide abrogates this effect restoring significantly (P < .01), in a dose-dependent manner, the fibrinolytic capability of endothelial cells to degrade the fibrin clot. HMECs indicates human dermal microvascular endothelial cells.
Defibrotide Protects ECs But not MM Cells Against Thalidomide Cytotoxic Effect
Two independent experiments have been performed to assess the influence of thalidomide and DF on the viability of cultured human microvascular ECs. First, colorimetric survival assays were carried out by incubating HMECs with increasing concentrations of thalidomide (0-100 µg/mL) and 150 µg/mL of DF. Figure 6, clearly demonstrated that exposure to increasing doses of thalidomide affects the in vitro viability of these cells in a dose-dependent manner. Importantly, a significant increase in survival was observed when activated HMECs were treated in the presence of 150 µg/mL DF showing a protection of thalidomide-induced cell death by DF (P < .01).
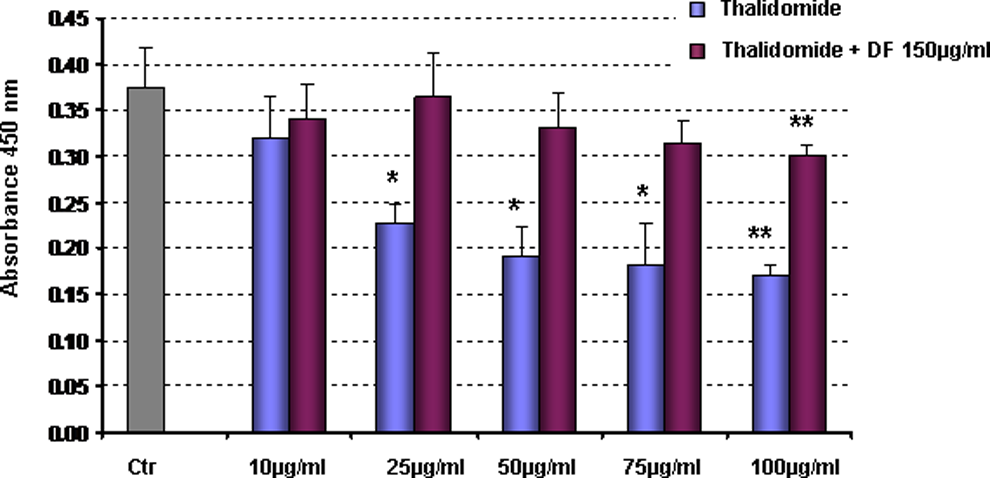
Influence of thalidomide and defibrotide on viability of cultured endothelial cells. HMECs were incubated with increasing concentration of thalidomide and defibrotide. MTT assay was carried out. Increasing doses of thalidomide significantly change, in a dose-dependent manner, the viability of HMEC in vitro (P < .01 andP < .05 thalidomide treatment vs control). Combined treatment with defibrotide protects the HMECs from thalidomide injury. HMECs indicates human dermal microvascular endothelial cells.
Furthermore, another experiment was performed for detecting apoptosis in HMECs using flow cytometric analysis. A concentration of 100 µg/mL thalidomide was chosen to stimulate HMECs in the presence or absence of 150 µg/mL DF. After 24 hours of incubation, HMECs were subjected to apoptosis assays using the detection of cellular granularity in PI-negative cells (SSC image in flow cytometry). Figure 7 demonstrated that thalidomide caused apoptosis in up to 60% of the HMEC cells, whereas DF protects the ECs against thalidomide cytotoxic effect.
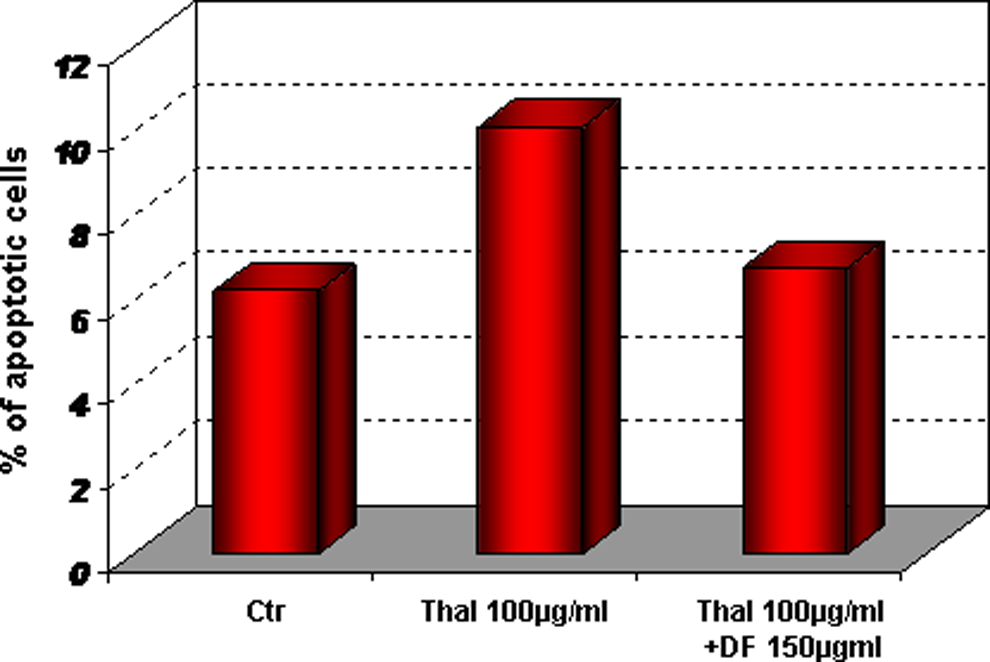
Defibrotide inhibits thalidomide-induced apoptosis in the HMECs. The cells were stimulated with thalidomide, in the presence or absence of defibrotide and subjected to flow cytometric assay. Thalidomide causes apoptosis in up to 60% of the cells; defibrotide protects the endothelial cells from apoptosis against thalidomide cytotoxic effect. HMECs indicates human dermal microvascular endothelial cells.
Apart from its desirable protective capacity for ECs, it was important to investigate whether DF could interfere with the antitumor properties of thalidomide. To address this question, MM cells were cultured with saline (control) or thalidomide (100 and 500 µg/mL) with and without 150 µg/mL DF for 24 hours. Figure 8 demonstrates a dose-dependency cytotoxicity of thalidomide in MM cells. In contrast to its protective effect on ECs, DF was unable to protect MM cells from thalidomide-mediated toxicity.
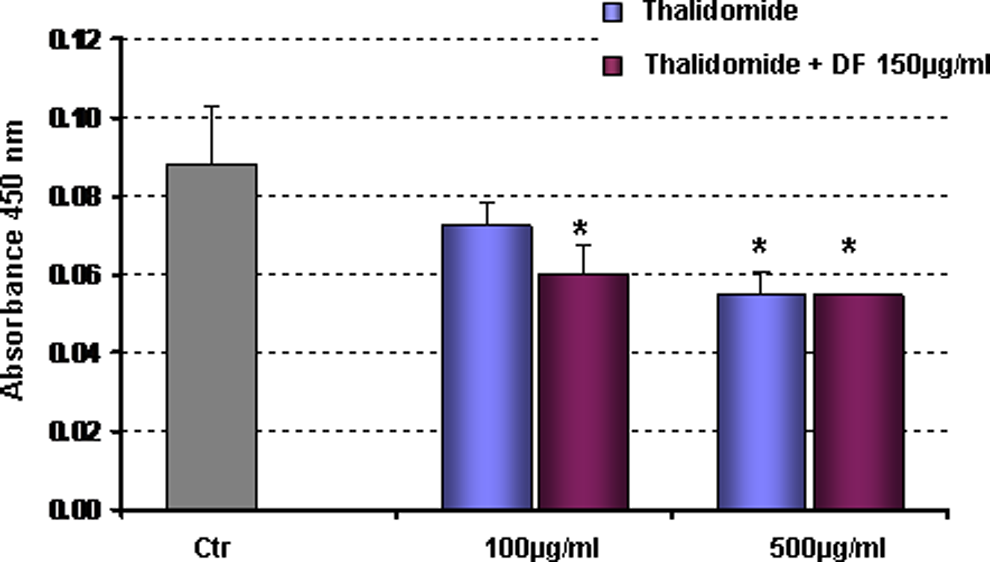
In vitro viability of MM cells treated with thalidomide or its combination with defibrotide. MM cells were stimulated with thalidomide in the presence and absence of defibrotide for 24 hours. After incubation, MTT assay was carried out to assess the impact of defibrotide on the in vitro response of MM cells to thalidomide. Defibrotide does not interfere with the thalidomide-mediated tumor cell toxicity. MM indicates multiple myeloma.
Similar results were observed with flow cytometric analysis. Figure 9 shows the apoptotic effect of thalidomide, whereas DF does not interfere with the desirable toxicity of thalidomide against MM cells.
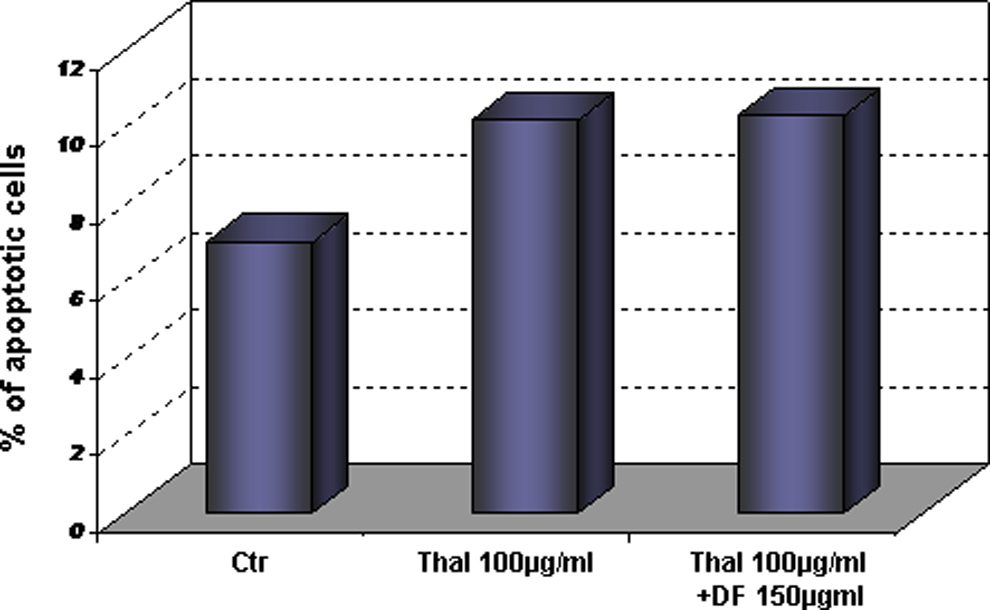
Defibrotide does not interfere with the antitumor effect of thalidomide. The cells were stimulated with thalidomide in the presence or absence of Defibrotide and subjected to flow cytometric assay. Thalidomide increases the percentage of apoptotic cells. Defibrotide was unable to protect MM cells from thalidomide-mediated toxicity. MM indicates multiple myeloma.
Discussion
Multiple myeloma is a malignant plasma cell disorder characterized by the accumulation of clonal plasma cells in the bone marrow, presence of a monoclonal immunoglobulin in serum and/or urine, and by evidence of end-organ damage including hypercalcemia, renal insufficiency, anemia, or lytic bone lesions. 19 Even with advances in therapeutic approaches, the disease remains incurable and is characterized by frequent relapses and fatal progression. However, the combination of novel biologically active agents with old chemotherapy shows promising results and changes the scenario of this disease. 20
Multiple myeloma is associated with an increase in risk of VTE. A baseline hypercoagulable state develops in MM, predisposing patients to a high risk of thromboembolism, especially when combined with chemotherapeutic and immunomodulatory drug agents such as thalidomide, mainly when used in combination therapy. 21
Despite the studies done in the past decade, the precise role of thalidomide therapy in the increased risk of TEEs remains difficult to establish because of the concurrent presence of many prothrombotic factors in patients with MM.
In a previous study designed to investigate the fibrinolytic activity in MM, Yağci et al established that patients with MM have a decreased fibrinolytic activity mainly because of a significant increase in plasma PAI-1 activity. 22
In our in vitro study, we demonstrated that MM cells, by themselves, alter the profibrinolytic potential of ECs. This effect was investigated by coculturing MM with HMEC cells. In this system, MM triggered a significant decrease in the release of t-PA and an induction in the expression of PAI-1 in ECs. We have also demonstrated that thalidomide potentiates this effect with a further decrease in the t-PA and induction of PAI-1 releases by ECs. Importantly, treatment with DF was able to restrain these events triggered by EC-MM cell interactions in the presence of thalidomide.
Additional analyses have been performed to verify whether the impact of DF in the fibrinolytic proteins (PAI-1 and t-PA) could also occur at transcriptional levels. The real-time results showed that thalidomide changes the gene expression of t-PA and PAI-1 in the HMEC cells, which is restored by DF.
The results of fibrinolytic assay showed that DF is able to counteract the modifications of antithrombotic phenotype in ECs modified by thalidomide increasing the fibrinolytic capacity of microvascular endothelium in a dose-dependent manner.
The profibrinolytic effect of DF has been suggested previously by others, and clinically DF has emerged as an active agent for the treatment and prevention of venous occlusive disease (VOD), a well-recognized, potentially life-threatening, rare complication following stem cell transplantation (SCT).23,24 In patients with VOD, DF has been shown to decrease PAI-1, increase t-PA levels, and increase the overall plasma fibrinolytic activity. 13 Similar results have been observed in preclinical studies in which DF dose dependently counteracted the increase in PAI-1 expression and decrease in t-PA activity consequent to LPS-induced endothelial damage. 8
Additionally, our in vitro results indicate that in addition to the effect of DF to counteract the modifications of antithrombotic phenotype of ECs induced by thalidomide, DF also protects ECs from thalidomide-mediated cell death.
The protective potential of DF has been previously demonstrated. Eissner et al showed that DF fully protected endothelial and epithelial cells from fludarabine (F-Ara)-mediated apoptosis. F-Ara, a nonmyeloablative immunosuppressant, has been shown to activate ECs causing damage when it is used in pharmacologically relevant concentrations. Defibrotide completely protected ECs from programmed cell death after the cotreatment with F-Ara, without affecting the antileukemic effect induced by the active metabolized form of fludarabine. 9
A key focus of this study was also to evaluate if next to the feature of DF to confer protection of endothelium cells from thalidomide injury, its administration could extend similar protective effects to neoplastic cells. Our present study indicates that DF does not counteract the thalidomide antitumor activity.
Similarly, Mitsiades et al demonstrated that DF does not blunt the anti-MM effect of several cytotoxic chemotherapy agents, such as DEX or the proteosome inhibitor bortezomib. Furthermore, DF increases in vivo sensitivity of MM cells to conventional chemotherapy by modulating the interactions of MM cells with their microenvironment. 15
Based on the preclinical results on MM models, the profibrinolytic and endothelial protective properties and safety profile of DF in other indications, such as prevention and treatment of VOD, a phase I/II study was conducted in patients with relapsed or relapsed/refractory MM treated with DF in combination with melphalan, prednisone, and thalidomide. Although the need of a further phase II study to better define the combination’s efficacy in large number of patients treated with the maximum tolerated doses of DF, Palumbo et al 25 demonstrated that the incidence of deep venous thrombosis (DVT) in patients treated with DF without any antithrombotic prophylaxis was only 4%. By comparison, DVT occurred in about 10% of patients receiving melphalan, prednisone, and thalidomide, despite the use of enoxaparin as anticoagulant prophylaxis. 26 Our data add to this work by showing that DF blunts the prothrombotic effect of thalidomide in ECs without adversely interfering with its antitumor effects. These findings can in part explain the fibrinolytic activity of DF and support its clinical use for the prevention of DVT induced by thalidomide or other immunomodulatory drugs.
Footnotes
This work has been presented orally at an open scientific session on the XXII Congress of the International Society on Thrombosis and Haemostasis, July 15, Boston, Massachusetts, USA.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.