
Abstract
Antiphospholipid antibodies contribute to the development of thrombosis, although precise mechanisms remain to be elucidated. We determined the effects of affinity-purified anti-beta2-glycoprotein 1 (anti-β2GP1) and anti-prothrombin (anti-PT) antibodies on in vitro platelet aggregation. Adenosine diphosphate (ADP) and collagen-induced platelet aggregation were performed using platelet-rich plasma ([PRP] 250 × 109/L). Antiphospholipid antibodies (1.25-10 μg/mL) were preincubated with PRP for 10 minutes at 37°C prior to the addition of agonist. Anti-β2GP1 antibodies significantly reduced platelet aggregation (percentage area under the curve; %AUC) in a concentration-dependent manner using both 5 μmol/L (P < .001) and 2.5 μmol/L (P = .038) ADP but did not significantly affect the rate of aggregation. Anti-PT antibodies significantly enhanced 5 µg/mL collagen-induced platelet aggregation (%AUC; P = .034) but did not affect ADP-induced platelet aggregation. These results suggest (1) interactions and effects of antiphospholipid antibodies on platelets are agonist and concentration dependent and (2) anti-β2GP1 antibodies may inhibit dense granule release and/or inhibition of the arachidonic acid pathway.
Introduction
Platelets help maintain hemostasis and are the first line of defense against loss of blood following trauma. This is achieved by adhesion, initial plug formation, activation, and aggregation. Activation causes the expression of anionic phospholipids on the platelet surface, the release of various hemostatic and nonhemostatic substances from platelet granules, and the initiation of the arachidonic acid pathway. The resulting production of thromboxane A2 (TxA2) in turn promotes further platelet activation.
Antiphospholipid antibodies are a heterogeneous group of immunoglobulins, including anti-cardiolipin (aCL) antibodies and lupus anticoagulants (LAs), which target phospholipids and phospholipid-binding proteins. The major protein targets of antiphospholipid antibodies are beta2-glycoprotein 1 (β2GP1) and prothrombin (PT), 1 although there are a wide range of other hemostatic proteins that have been described and investigated. 2–5
Patients with antiphospholipid antibodies, for example, the antiphospholipid syndrome (APS) and systemic lupus erythematosus (SLE), are at a higher risk of thrombosis, 6,7 although precise mechanisms remain unclear. Indeed, there are conflicting reports on the interactions between antiphospholipid antibodies and platelets. It has previously been reported that platelet hyperfunction (as measured by platelet aggregation) is present in patients with antiphospholipid antibodies, 8 suggesting an important role for platelets in the development of thrombosis in these patients. Furthermore, it has been demonstrated that aCL and/or LA from patients with SLE bind to several platelet membrane proteins, for example, platelet glycoprotein IIIa, suggesting that antiphospholipid antibodies may contribute to mediating platelet activation. 9,10 In contrast, prolonged bleeding times (as a marker of primary hemostasis), but without a bleeding phenotype, have been reported in patients with antiphospholipid antibodies, 11,12 instead suggesting these antibodies dampen rather than increase platelet function.
Thus, the aim of this study was to investigate the effects of the antiphospholipid antibodies, anti-β2GP1 and anti-PT, on platelet activation and aggregation, using in vitro platelet aggregation.
Materials and Methods
Ethics
This study was approved by the Tasmanian Health and Medical Human Research Ethics Committee (Reference number H009415).
Sample Preparation and Processing
Whole blood (9 parts) from healthy volunteers was collected in 3.8% sodium citrate (1 part) with minimal stasis using vacutainers (Vacuette, Greniner Bio-One, GmbH, Austria). All volunteers had not taken aspirin or other antiplatelet medications in the previous 10 days and had avoided foods known to affect platelet aggregation, consistent with the previous work of our group. 13 Platelet-rich plasma (PRP) was obtained following centrifugation for 10 minutes at 150g at 20°C. Platelet-poor plasma (PPP) from the same samples was prepared following further centrifugation for 20 minutes at 2000g at 20°C. Platelet counts were performed on PRP using a blood analyser (Sysmex XS 1000i, Sysmex America Inc, Mundelein, IL, USA) and adjusted to 250 × 109/L for each experiment using PPP from the same normal donor. Testing of platelets was completed within 3 hours.
Effect of Antiphospholipid Antibodies on Platelet Aggregation
Platelet aggregation using light transmission aggregometry was performed using a 4-channel AggRAM module (Helena Laboratories, Beaumont, TX, USA). Adenosine diphosphate (Sigma-Aldrich, Sydney, Australia) and equine collagen (Helena Laboratories, Beaumont, TX, USA) were used as agonists. 200 μL of PRP was incubated for 10 minutes at 37°C with 25 μL of affinity-purified polyclonal anti-β2GP1 or anti-PT (rabbit anti-human antibodies; Hyphen-Biomed, Neuville-Sur-Oise, France; final concentrations 10 to 1.25 μg/mL). All experiments were performed in duplicate and in parallel with a duplicate of PRP incubated with 25 μL of buffered normal saline ([BNS] pH 7.1) that was used as the baseline. Aggregation was initiated upon the addition of 25 μL of agonist (5 or 2.5 μmol/L ADP; 5 or 2.5 μg/mL collagen; final concentrations). Parameters including percentage area under the curve (%AUC), slope, and percentage maximum aggregation (%Max) of samples were generated by the aggregometer software (HemoRAM 1.1.0., Helena Laboratories). Test results were compared to the baseline control run in conjunction with each sample and results for all parameters measured were recorded as the mean difference between the test and baseline. Platelet-rich plasma used for test and baseline control runs were from the same donor for each experiment.
Statistical Analysis
All figures were generated using GraphPad Prism 5 (Intuitive Software for Science, San Diego, California). STATA version 10.0 (Statacorp LP, College Station, TX, USA) was used to analyze platelet aggregation with a general linear model to test for any differences between the baseline parameters and the antibody results. Postestimation Holm test analysis was then used to adjust P values for multiple comparisons. Results for platelet aggregation are expressed as the mean difference from baseline ± standard error of the mean (SEM). For all analyses P values less than .05 were considered statistically significant.
Results
Results for %AUC and %Max were similar for all experiments, thus only %AUC data are presented.
Effect of Antiphospholipid Antibodies on ADP-Induced Platelet Aggregation
Anti-β2GP1 antibodies inhibited ADP-induced aggregation in a concentration dependent manner (typical platelet aggregation traces are presented in Figure 1 ). Anti-β2GP1 antibodies significantly reduced %AUC in a concentration-dependent manner when incubated with normal platelets induced by both 5 μmol/L and 2.5 μmol/L ADP (P < .001 and P = .038; Figure 2A and B). There was no significant difference in the primary slope of aggregation for platelets incubated with anti-β2GP1 antibodies (P> .05; Figure 2C and D). There was no significant difference or concentration-dependent response in platelet aggregation (both %AUC and slope) in platelets incubated with anti-PT antibodies when induced by either 5 µmol/L or 2.5 μmol/L ADP (Figure 3 ).
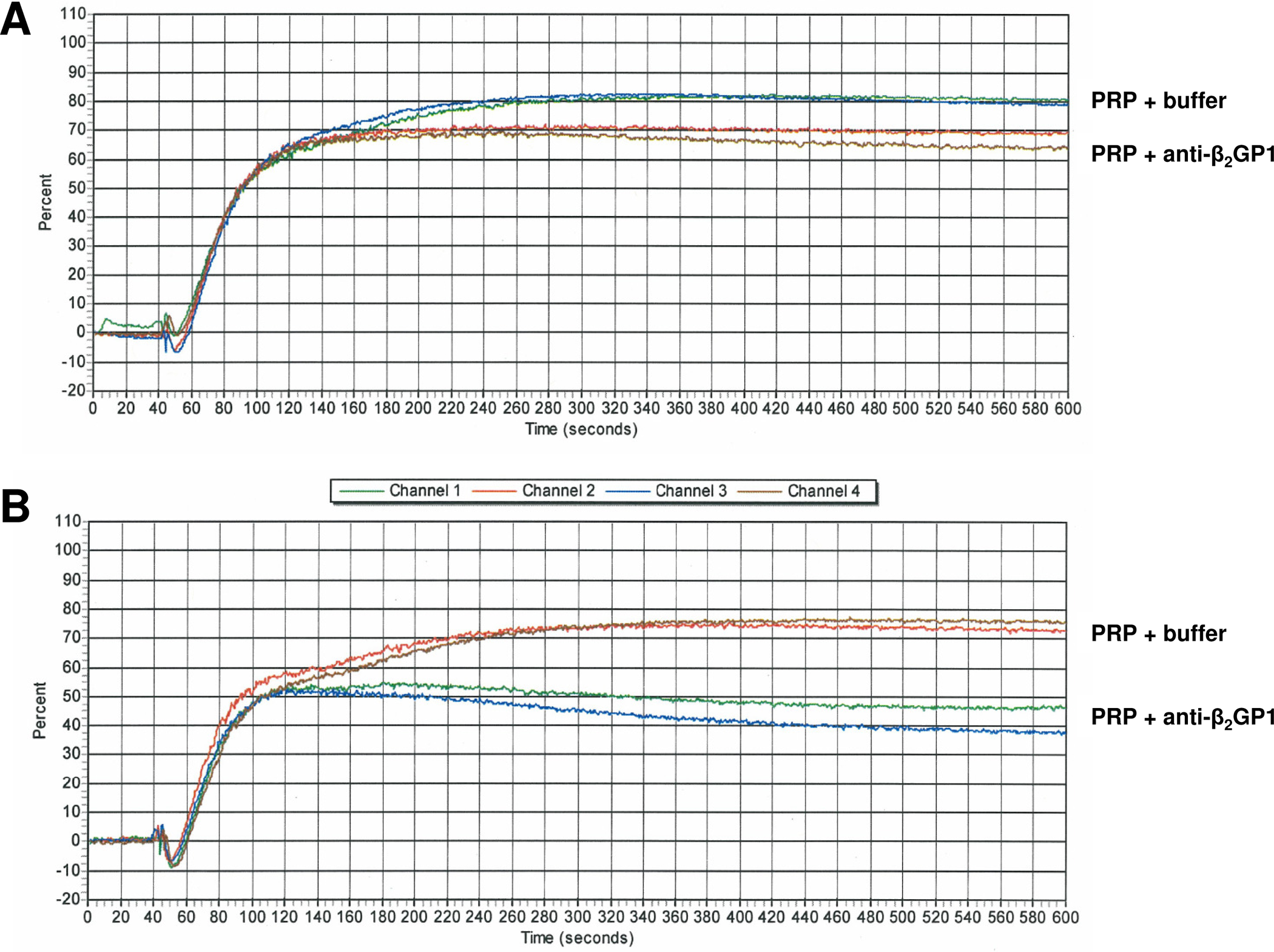
Representative traces demonstrating the effect of anti-beta2-glycoprotein 1 (anti-β2GP1) antibodies on ADP-induced platelet aggregation. Channels 1 and 3: platelet-rich plasma (PRP) + 10 μmol/L anti-β2GP1. Channels 2 and 4: PRP + buffer. A, 5 μmol/L adenosine diphosphate (ADP). B, 2.5 μmol/L ADP.
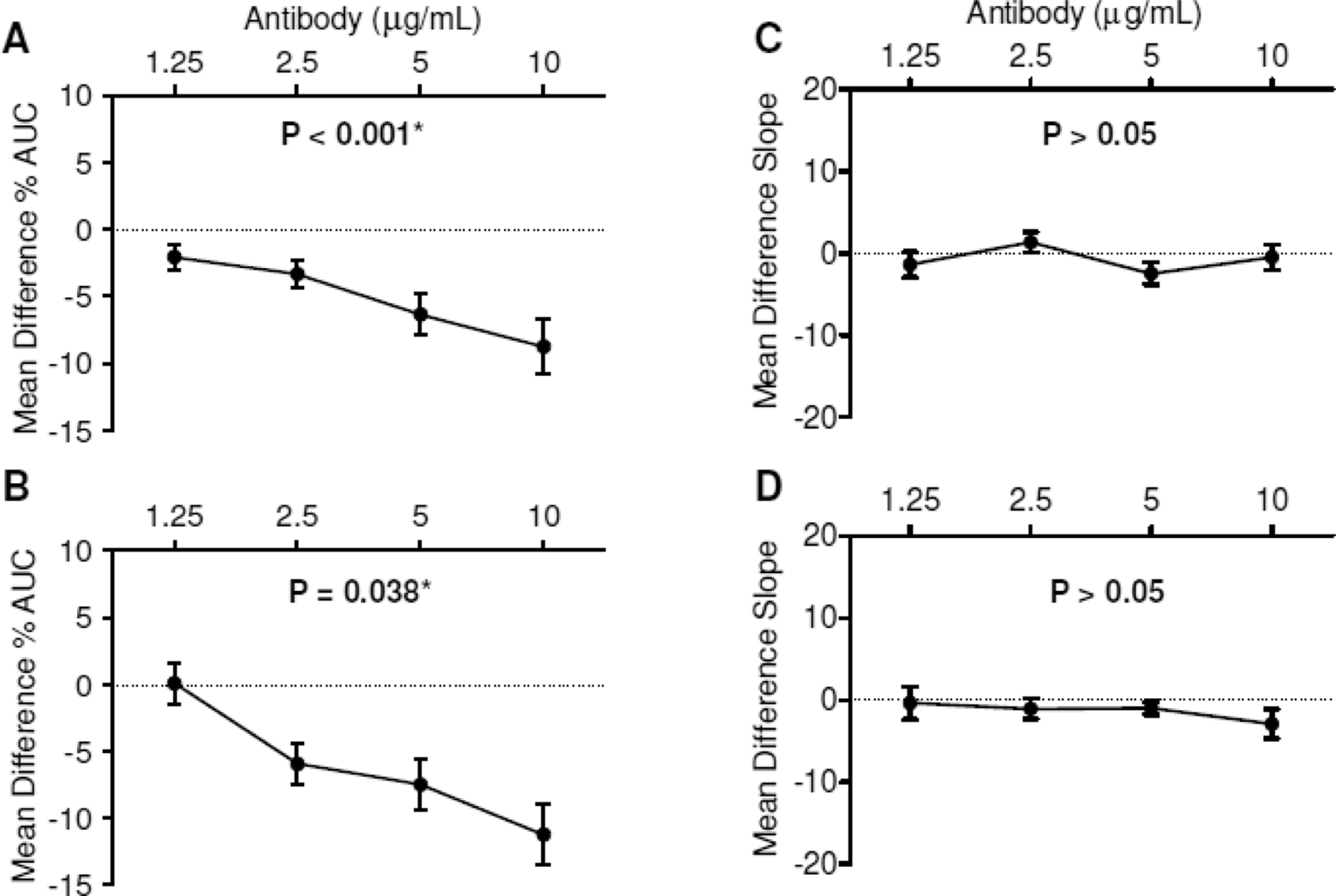
Effect of anti-beta2-glycoprotein 1 (anti-β2GP1) antibodies on adenosine diphosphate (ADP)-induced platelet aggregation. Dotted line at mean difference %Max = 0 represents baseline (platelet-rich plasma [PRP] with only buffer added). Percentage area under the curve (%AUC) was significantly inhibited by anti-β2GP1 antibodies in a concentration-dependent manner. A, 5 μmol/L adenosine diphosphate (ADP). B, 2.5 μmol/L ADP. The rate of aggregation (slope) was not significantly affected by anti-β2GP1 antibodies. C, 5 μmol/L ADP. D, 2.5 μmol/L ADP. Data presented as mean ± standard error of the mean ([SEM] n = 12-23).
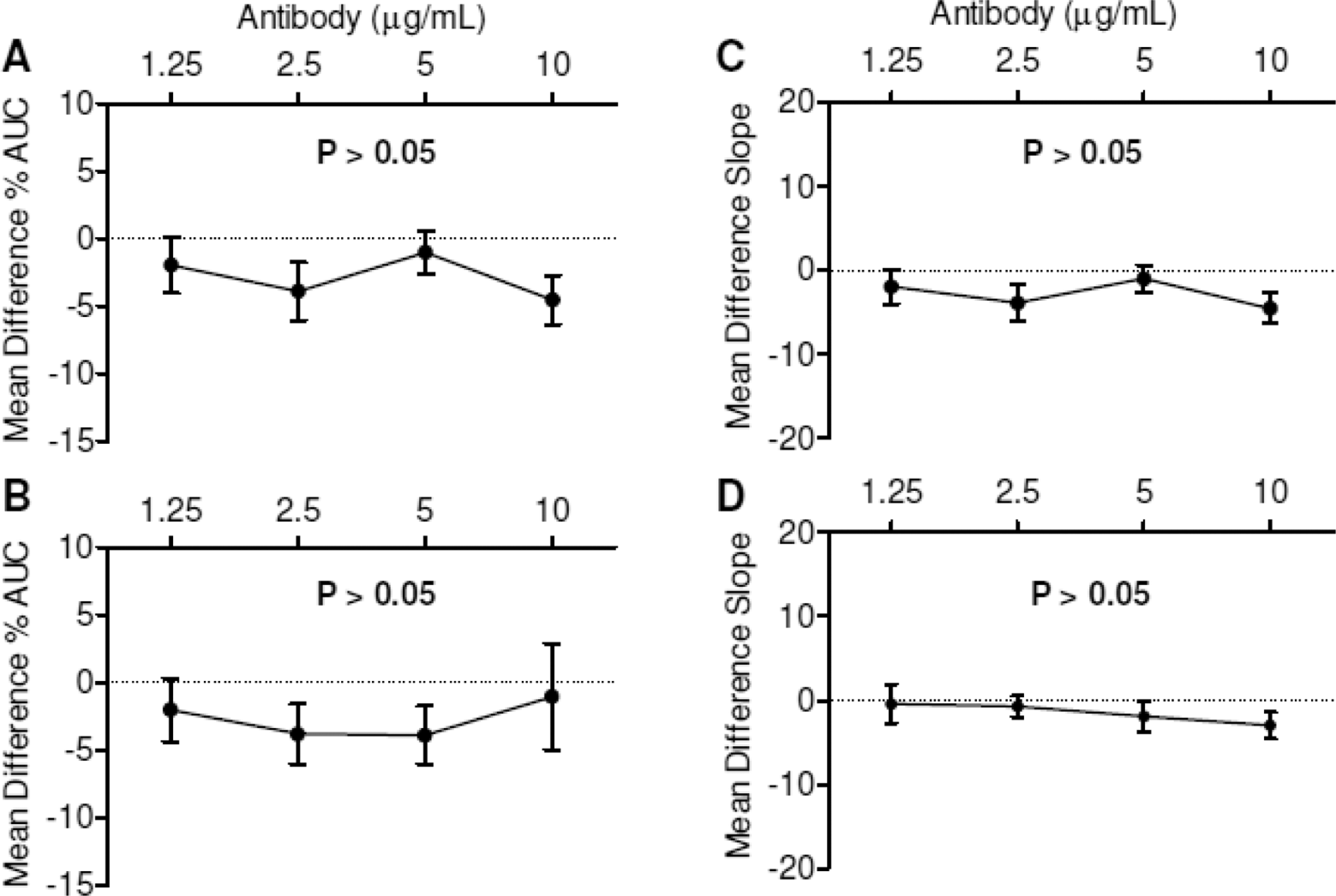
Effect of anti-prothrombin (anti-PT) antibodies on adenosine diphosphate (ADP)-induced platelet aggregation. Dotted line at mean difference percentage maximum aggregation (%Max) = 0 represents baseline (platelet-rich plasma [PRP] with only buffer added). Percentage area under the curve (%AUC) and slope were not significantly altered by anti-PT antibodies. A and C, 5 μmol/L ADP. B and D, 2.5 μmol/L ADP (n = 10-21).
Effect of Antiphospholipid Antibodies on Collagen-Induced Platelet Aggregation
Anti-β2GP1 antibodies significantly enhanced %AUC induced by 2.5 µg/mL (P = .018; Figure 4B) but not 5 µg/mL (Figure 4A) collagen. There was a significant difference in the rate of aggregation (secondary slope) using 2.5 µg/mL (P = .018; Figure 4D) but not 5 µg/mL (Figure 4C) collagen. Anti-PT antibodies significantly enhanced %AUC when induced by 5 µg/mL (P = .028; Figure 5A) but not 2.5 µg/mL (Figure 5B) collagen. There was a significant difference in the rate of aggregation (secondary slope) using 2.5 µg/mL (P = 0.018; Figure 5D) but not 5 µg/mL (Figure 5C) collagen.
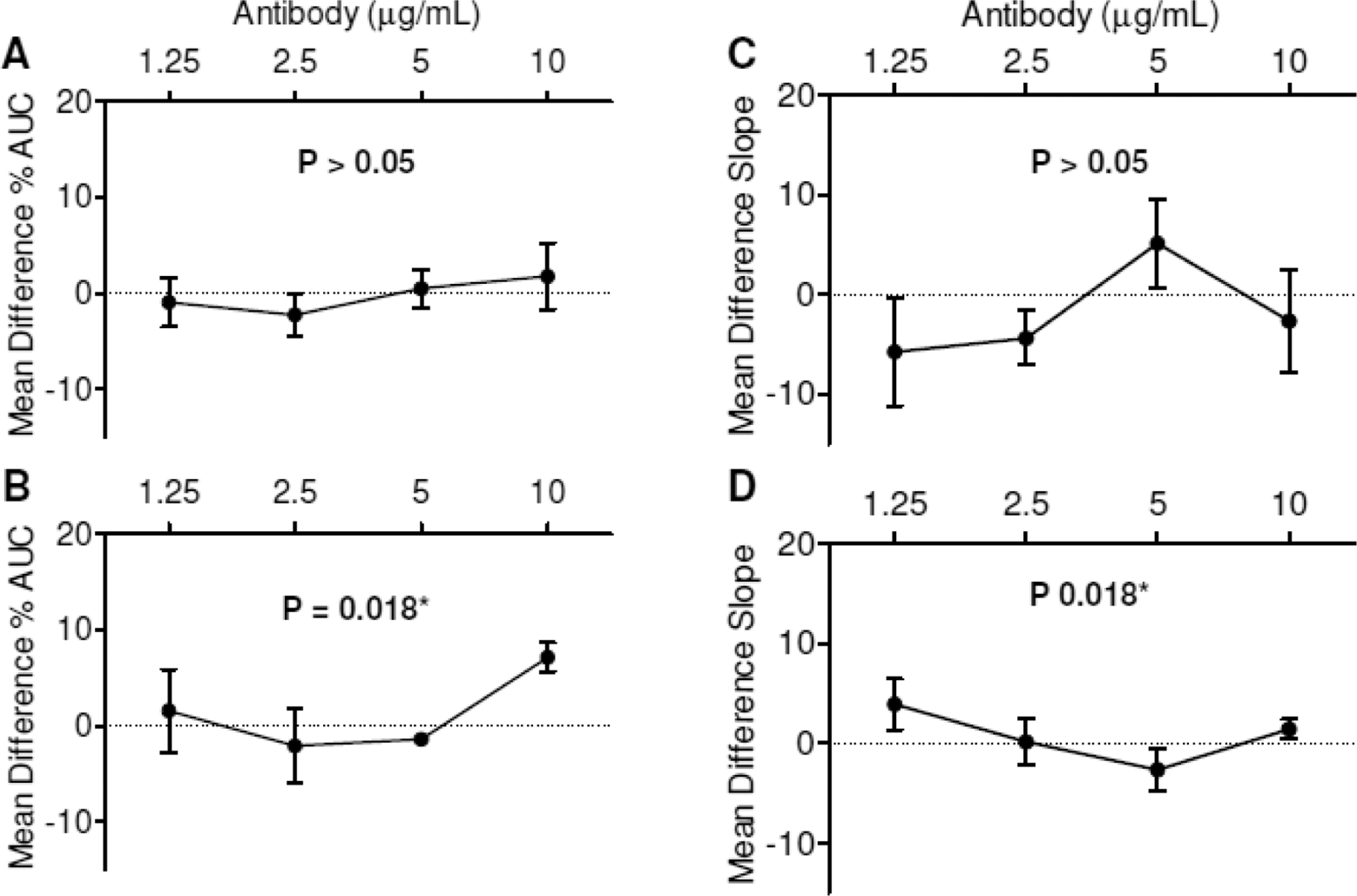
Effect of anti-beta2-glycoprotein 1 (anti-β2GP1) antibodies on collagen-induced platelet aggregation. Dotted line at mean difference percentage maximum aggregation (%Max) = 0 represents baseline (platelet-rich plasma [PRP] with only buffer added). Percentage area under the curve (%AUC; 2.5 µg/mL but not 5 µg/mL collagen) was significantly enhanced by anti-β2GP1 antibodies. A, 5 µg/mL collagen. B, 2.5 µg/mL collagen. Similarly, the rate of aggregation (slope; 2.5 µg/mL but not 5 µg/mL collagen) was significantly enhanced by anti-β2GP1 antibodies. C, 5 µg/mL collagen. D, 2.5 µg/mL collagen (n = 3).
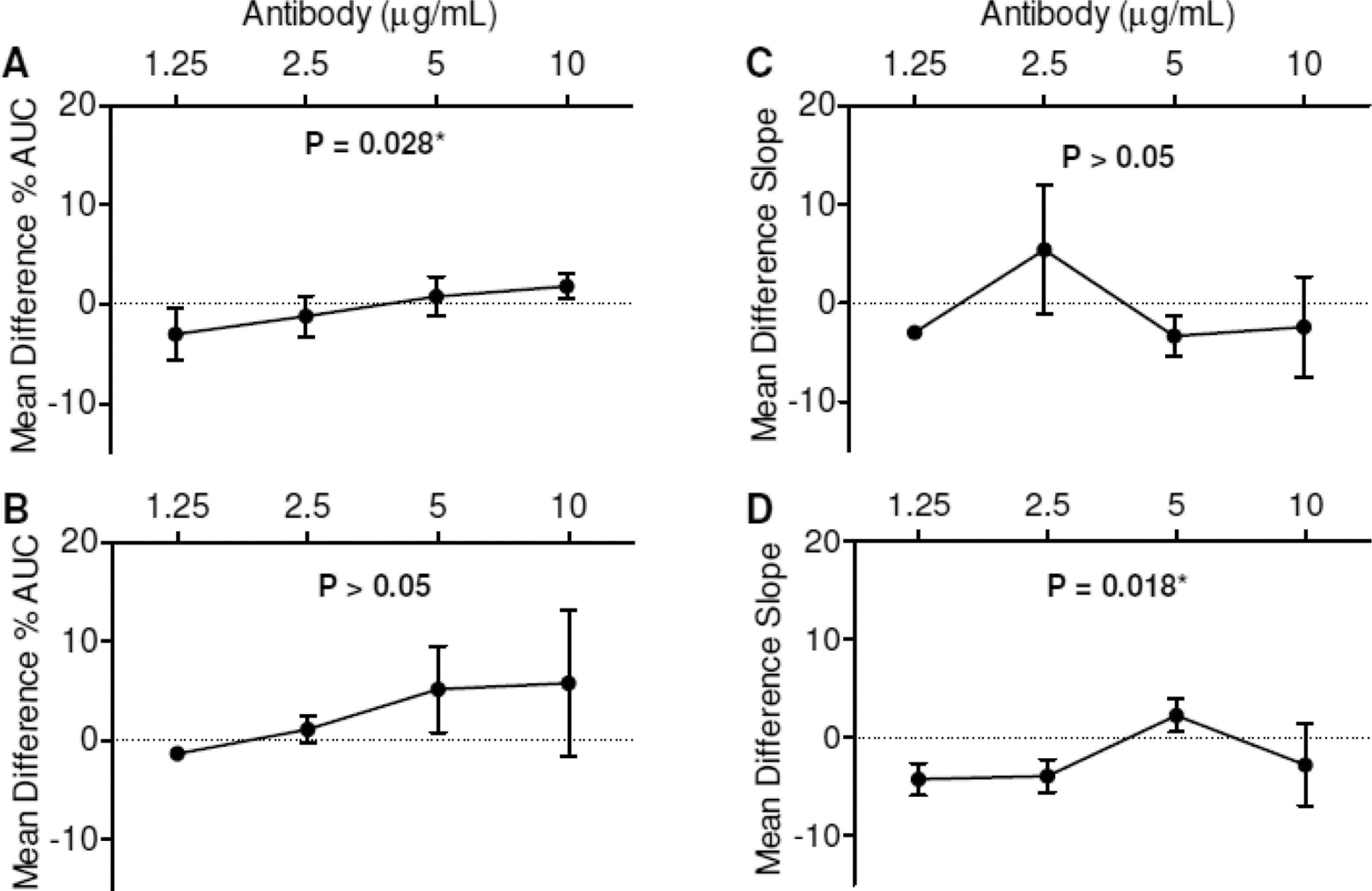
Effect of anti-prothrombin (anti-PT) antibodies on collagen-induced platelet aggregation. Dotted line at mean difference percentage maximum aggregation (%Max) = 0 represents baseline (platelet-rich plasma [PRP] with only buffer added). Percentage area under the curve (%AUC; 5 µg/mL but not 2.5 µg/mL collagen) was significantly enhanced by anti-PT antibodies. A, 5 µg/mL collagen. B, 2.5 µg/mL collagen. The rate of aggregation (slope) was not significantly affected by anti-PT antibodies. C, 5 µg/mL collagen. D, 2.5 µg/mL collagen (n = 3).
Discussion
This study demonstrated that affinity-purified anti-β2GP1 and anti-PT have variable effects on in vitro platelet aggregation. The effects were dependent on the type and concentration of agonist used, as well as the type of antiphospholipid antibody. Furthermore, it was clearly and consistently demonstrated that anti-β2GP1 antibodies significantly reduced in vitro ADP-induced platelet aggregation in a concentration-dependent manner.
It is interesting to speculate how ADP-induced platelet aggregation may be inhibited by anti-β2GP1 antibodies. ADP induces platelet aggregation by binding to the G-protein-coupled receptors, P2Y1 and P2TAC, that are located on platelet membranes. 14 This leads to the inhibition of adenyl cyclase, exposure of fibrinogen receptors, and the activation of phospholipase enzymes that metabolize phosphatidylinositol to activate the arachidonic acid pathway. Direct inhibition of these receptors may potentially impede phospholipase C production, leading to decreased arachidonic acid and TxA2 production. The results from our study, however, demonstrate that the rate (slope) of aggregation was not affected, suggesting that anti-β2GP1 antibodies do not affect primary aggregation but rather impede the secondary aggregation mechanism. This is consistent with previous work that has demonstrated ADP-induced primary aggregation occurs independently of phospholipase C-induced phosphatidylinositol metabolism. 15,16 It may instead be that anti-β2GP1 antibodies inhibit secondary aggregation by impairing the release of dense granule contents, for example, ADP, adenosine triphosphate (ATP), and serotonin, as well as TxA2 production, during anti-β2GP1-spiked ADP-induced platelet aggregation.
A further explanation for the data in the present study may be that anti-β2GP1 antibodies enhance the modulating effect of its target protein, β2GP1. It has previously been reported that a possible in vitro function of β2GP1 is the inhibition of ADP-induced aggregation, by interfering with the release reaction of dense granules. 17 It was demonstrated that concentrations of β2GP1 greater than 0.1 mg/mL inhibited serotonin release from platelets using ADP concentrations from 2 μmol/L to 10 μmol/L but did not affect collagen- or thrombin-induced aggregation. Whether the binding of anti-β2GP1 antibodies to β2GP1 inhibits the release of other important platelet activation agents, such as ADP and calcium ions, from platelet-dense granules remains to be determined.
In contrast to the ADP results, the effects of antiphospholipid antibodies on collagen-induced platelet aggregation were significantly more variable. Collagen stimulates irreversible in vitro platelet aggregation by binding to glycoprotein membrane receptors Ia/IIa (GP Ia/IIa) and VI (GP VI), inducing changes in shape and exposure of membrane phospholipids. 18 Low concentrations of collagen (1-2 μg/mL) stimulate platelet aggregation through mechanisms dependent on the arachidonic pathway and subsequent TxA2 synthesis. Alternatively, high concentrations of collagen can induce aggregation by directly increasing calcium concentrations, thus releasing the contents of alpha and dense granules. How collagen-induced platelet aggregation was enhanced by both anti-β2GP1 and anti-PT antibodies used in this study is unclear, however they may bind to collagen-dependent receptors to stimulate further aggregation. It has previously been proposed that complexes of β2GP1-anti-β2GP1 may bind to negatively charged phospholipids that become exposed following activation and interact with the apolipoprotein ER2 receptor, resulting in platelets becoming more sensitive to the effects of collagen and subsequent platelet activation. 19 It may also be possible that complexes of PT dimers, PT-anti-PT, or anti-PT alone may interact with collagen-dependent mechanisms, where it has been reported that PT dimers have increased affinity for phospholipids. 20
It has been well described that antiphospholipid antibodies, particularly anti-β2GP1, are associated with hypercoagulability and thrombotic complications. 19–22 The results from our present study instead suggest that anti-β2GP1 antibodies may reduce hypercoagulability by inhibiting ADP-induced platelet aggregation. It is known that a significant proportion of patients with antiphospholipid antibodies demonstrate a prolonged bleeding time (reflecting impaired platelet function) without an accompanying bleeding phenotype, 11,12 thus it may be that anti-β2GP1 antibodies play an important role in dampening the platelet response in at least a proportion of these patients.
In conclusion, this study demonstrated that affinity-purified antiphospholipid antibodies, anti-β2GP1 and anti-PT, have variable effects on in vitro platelet aggregation. It is clear that anti-β2GP1 antibodies generate a concentration-dependent inhibitory effect that reduces overall (%AUC), but not the rate (slope) of the primary wave of ADP-induced aggregation. Further studies are warranted to determine the precise interactions and thus the role of antiphospholipid antibodies in platelet function. Whether the in vitro effects demonstrated here are also significant in vivo remains to be determined. Finally, further investigation into the response of platelets to other agonists such as thrombin, TxA2, and arachidonic acid may provide further insight into the direct mechanisms involved in antiphospholipid antibody interactions with platelets.
Footnotes
Acknowledgments
We would like to thank Dr Iain Robertson for advice with statistical analysis, as well as Merrilyn Johnson for her excellent technical assistance.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article:This study was part funded by the Clifford Craig Medical Research Trust and by a Faculty of Health Science Early Career Research Funding Award (for MJA) from the University of Tasmania.