
Abstract
Introduction
The development of safe and effective hemostatic adjuncts is important to reduce the time required for surgeons to achieve hemostasis and to minimize the incidence of bleeding-related complications. Fibrin sealants typically contain fibrinogen and thrombin, which are delivered simultaneously to a bleeding site to achieve hemostasis. 1 The hemostatic effectiveness and safety of fibrin sealants have been previously demonstrated in a wide variety of surgical procedures, spanning multiple surgical specialties.2–9
Recent controversy surrounding the safety of an antifibrinolytic agent, aprotinin, has prompted review of its use. 10 Aprotinin and tranexamic acid, which are both antifibrinolytic agents, may not be necessary in the formulation of effective fibrin sealants. The elimination of these agents may enhance the safety profile and allow for wider use of fibrin sealants, particularly in neurosurgical procedures.
A phase III, randomized, single-blind, multicenter study 2 demonstrated that an aprotinin-free fibrin sealant significantly reduced the time to hemostasis during hepatic resection, when compared with conventional hemostatic techniques. The fibrin sealant evaluated in the current study is a new version of that fibrin sealant; because its manufacturing process includes a step for removal of plasminogen, the new formulation does not contain tranexamic acid or aprotinin. In addition, results of animal studies indicate that the hemostatic activity and clot longevity associated with the new formulation of fibrin sealant are similar to those of its predecessor. 11 The current study evaluated the effectiveness and safety of this tranexamic acid- and aprotinin-free fibrin sealant compared with an absorbable hemostat in achieving hemostasis in soft tissue during elective retroperitoneal or intra-abdominal surgery.
Methods
Patients
This phase III, randomized, active-controlled, multicenter study was conducted at 16 US surgical centers between February 28, 2006, and December 27, 2006. Approval was obtained from the Institutional Review Board at each participating site. The study was conducted under the US Food and Drug Administration Investigational New Drug regulations (21 CFR 312), International Conference on Harmonisation (1996), and the Declaration of Helsinki (1996).
Patients who were scheduled to undergo elective retroperitoneal or intra-abdominal surgery were screened within 21 days of surgery to determine their eligibility for study enrollment. Patients were excluded from the study if they were undergoing an emergent procedure, were unwilling to receive blood products, had known intolerance to blood products or to one of the components of the study product, had an autoimmune immunodeficiency disease, had a history of alcohol or drug abuse, were pregnant or nursing, or had participated in another investigational drug or device research study within 30 days of enrollment.
A signed informed consent form was obtained from patients who were willing to participate in the study. For those patients younger than 18 years, consent was obtained from the patient’s parent or legal guardian.
Patients who were eligible for enrollment underwent surgical procedures according to the institution’s standard of care. Once the surgeon encountered the first challenging soft-tissue bleeding as a result of dissection at the site of the primary operative intent, conventional surgical techniques (sutures, ligature, cautery) were employed to attempt to control the bleeding at the target bleeding site (TBS). If the use of these conventional techniques was considered to be impractical or ineffective, the degree of bleeding at the TBS was recorded (mild bleeding defined as small areas of capillary, arteriole, or venule oozing in tissues; moderate bleeding defined as either a larger area of bleeding of a magnitude similar to that described for mild bleeding or bleeding that was more pronounced, such as flowing or pulsatile, but that was not massive or from a large artery). Patients were then assigned 1:1 to a treatment group by opening a randomization envelope provided by the study sponsor, with randomization balanced within each study site. The time at which the randomization envelope was opened was defined as 0 minutes (T0). Bleeding was assessed at 4, 7, and 10 minutes following randomization, with hemostasis defined as the absence of detectable bleeding at the TBS. Time was recorded by an independent observer with a stopwatch, while the surgeon called out surgical events related to the TBS.
Interventions
Patients received either fibrin sealant (EVICEL* Fibrin Sealant [Human], Johnson & Johnson Wound Management, a Division of ETHICON, Inc, Somerville, New Jersey) or absorbable hemostat (SURGICEL Absorbable Hemostat, Johnson & Johnson Wound Management, a Division of ETHICON, Inc, Somerville, NJ) as an adjunct for hemostasis. Fibrin sealant and the absorbable hemostat were both prepared in advance (ie, prior to the identification of the TBS and prior to randomization) and were ready to use in the operating room for each patient. The nonassigned treatment was removed from the operating room immediately following randomization to a treatment group. Fibrin sealant was administered by either dripping or spraying onto the TBS of patients who were randomized to receive fibrin sealant. The absorbable hemostat was directly applied to the TBS of patients randomized to this treatment group, and manual compression was applied.
Main Outcome Measures
The primary effectiveness endpoint was the hemostasis outcome at 10 minutes, with success defined as the absence of bleeding and absence of treatment with additional hemostatic measures. Secondary effectiveness endpoints included the hemostasis outcome at 4 and 7 minutes, the incidence of any complications that were potentially related to bleeding from the time of randomization to follow-up (7-14 days following the procedure), and the incidence of treatment failures (defined as the presence of bleeding at the TBS 10 minutes postrandomization or brisk bleeding that required the use of additional hemostatic measures during the 10-minute observation period). The absolute time to hemostasis (TTH) was also recorded for each patient. Adverse events were recorded as they were reported from the time of randomization to the time of follow-up 7 to 14 days after surgery.
Statistical Analyses
For the purpose of sample size calculations, it was assumed that the proportion of patients who would receive fibrin sealant or an absorbable hemostat and achieve hemostasis within 10 minutes would be 0.9.12,13 It was also assumed that the TTH is similar in retroperitoneal or intra-abdominal surgery and that the fibrin sealant would be considered noninferior to the absorbable hemostat if the lower limit of the 2-sided 95% confidence interval (CI) for the relative risk was more than 0.80. Using these assumptions and the method described by Farrington and Manning, 14 a total sample size of 126 patients who would complete the study would be needed to achieve 95% confidence and 90% power; this sample size was increased to 130 patients to allow for early study discontinuations.
The intent-to-treat population, which included all adult patients who were randomized to a treatment group, was used for analyses of effectiveness measures. Safety measures were analyzed using all adult patients who were randomized and received treatment with fibrin sealant or the absorbable hemostat (safety population). Pediatric patients (<17 years of age) were not included in these analyses because of the low sample size.
A 2-sided Koopman 15 95% CI was constructed to evaluate the ratio of 2 proportions in effectiveness endpoints between the treatment groups. This method is consistent with the chi-square test for 2 × 2 tables. If the lower limit of the 95% CI was more than 0.80, the fibrin sealant was determined to be noninferior to the absorbable hemostat. If the lower limit was more than 1.0, this was considered evidence for the superiority (P < .05) of the fibrin sealant over the absorbable hemostat, and a P value would be calculated using a logistic model with treatment and age group as variables. The overall distribution of patients with mild or moderate bleeding between the 2 treatment groups was evaluated using a chi-square test.
Results
A total of 124 adult patients were enrolled and randomized to receive fibrin sealant (n = 62) or absorbable hemostat (n = 62) as an adjunct for hemostasis. The flow of patients through the trial is illustrated in Figure 1 . Demographic and baseline characteristics were similar between the treatment groups (Table 1 ). For most patients, bleeding at the TBS was considered to be mild (fibrin sealant, n = 39 [62.9%]; absorbable hemostat, n = 36 [58.1%]), and for the remainder of patients, bleeding was moderate (fibrin sealant, n = 23 [37.1%]; absorbable hemostat, n = 26 [41.9%]); there was no statistical between-group difference in the severity of bleeding at the TBS (P = .5831).
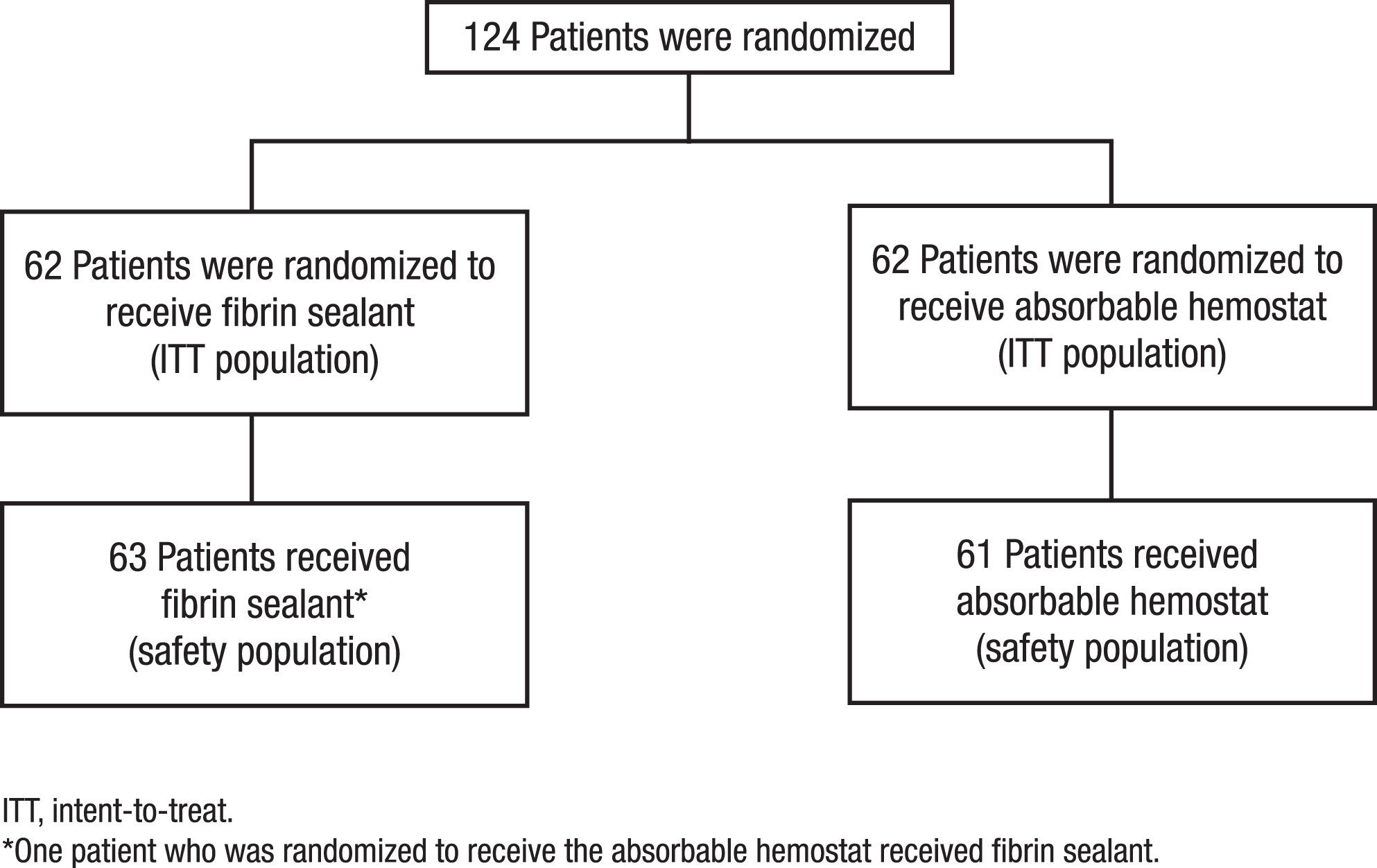
Flow of patients through the study.
Demographic and Baseline Characteristics
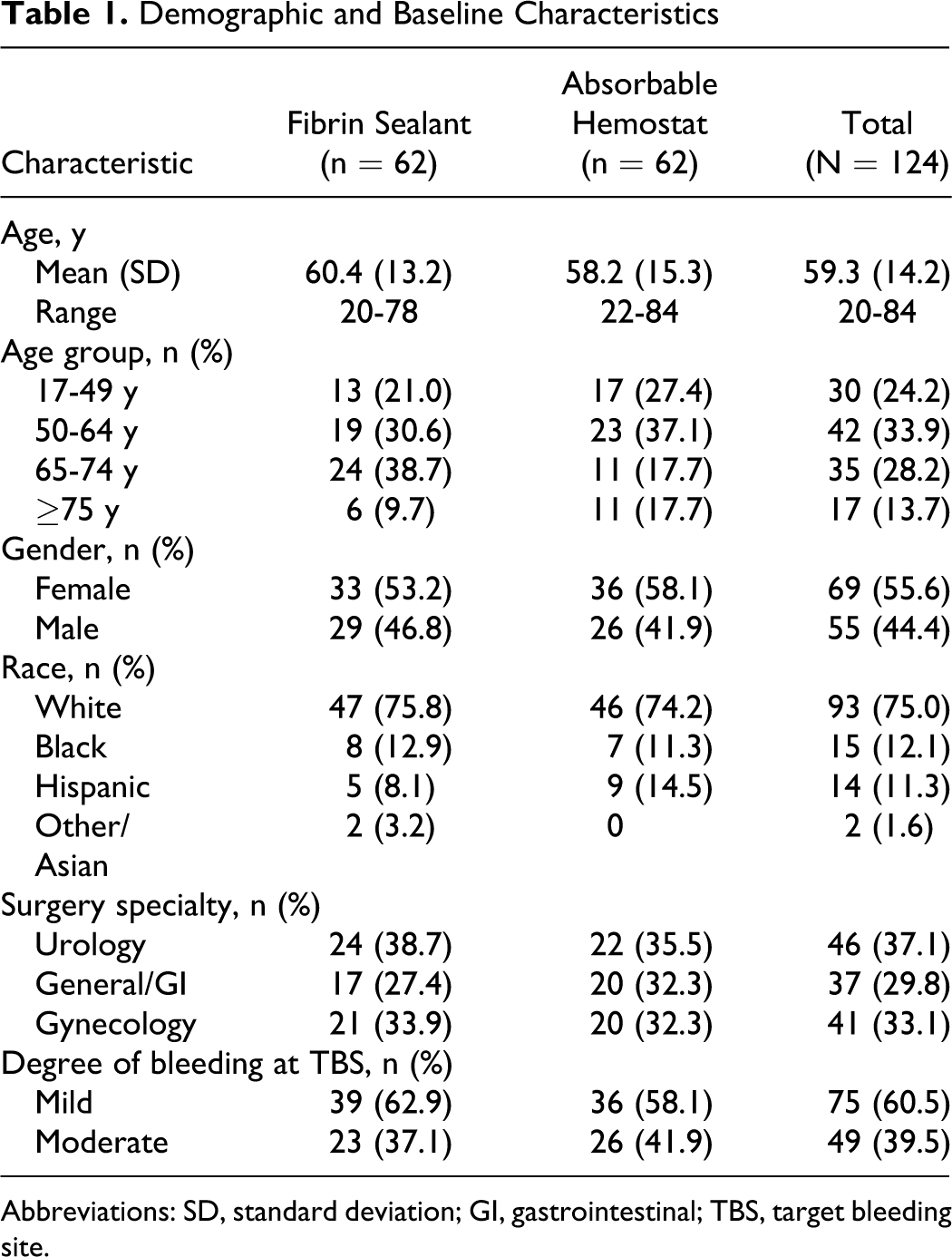
Abbreviations: SD, standard deviation; GI, gastrointestinal; TBS, target bleeding site.
The distribution of surgical procedures was similar between groups (Table 1). Urologic surgical procedures included simple or radical nephrectomy, radical cystectomy, adrenalectomy (open), radical prostatectomy, and pyeloplasty. Gynecologic surgical procedures included radical hysterectomy, lymphadenectomy, and primary tumor reduction surgery (ie, ovarian cancer surgery). General surgical procedures included colectomy with or without anal anastomoses, low anterior resection, abdominoperineal resection, and retroperitoneal tumor resection. The median duration of surgery was 145 minutes (range, 31-831 minutes) for patients who received fibrin sealant and 157 minutes (range, 34-822 minutes) for patients who received the absorbable hemostat.
A higher percentage of patients who received fibrin sealant achieved hemostasis at 10 minutes compared with patients who received the absorbable hemostat (relative risk = 1.16; 95% CI, 1.02-1.35; P < .05; Figure 2 ). A higher percentage of patients receiving fibrin sealant versus absorbable hemostat also achieved hemostasis at the 4-minute (relative risk = 1.35; 95% CI, 1.04–1.80; P < .05) and 7-minute (relative risk = 1.17; 95% CI, 1.00-1.39; P = .05) time points (Figure 3 ).
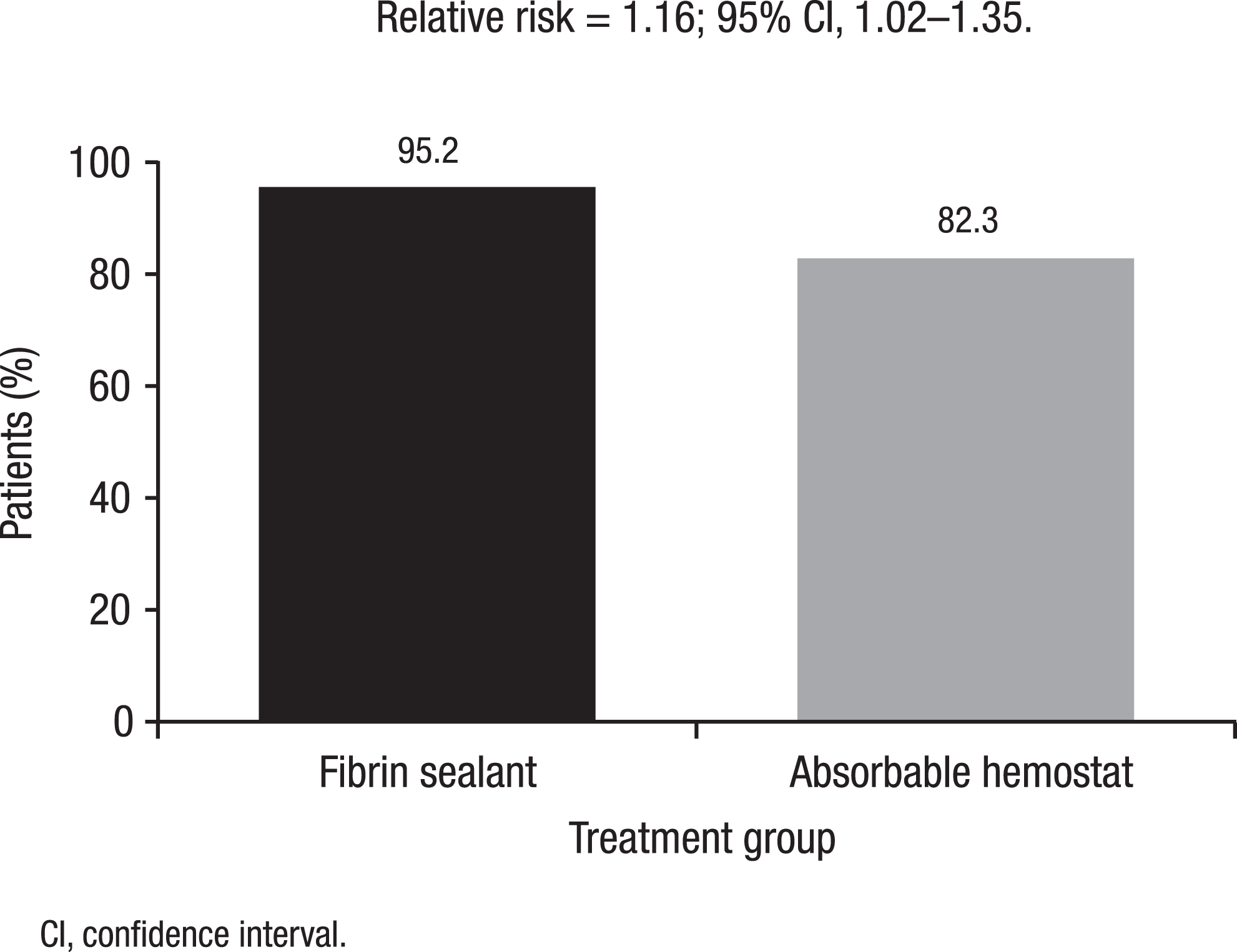
Percentages of patients who achieved hemostasis within 10 minutes. The percentages of patients who received fibrin sealant or the absorbable hemostat and achieved hemostasis at the target bleeding site within 10 minutes are given.
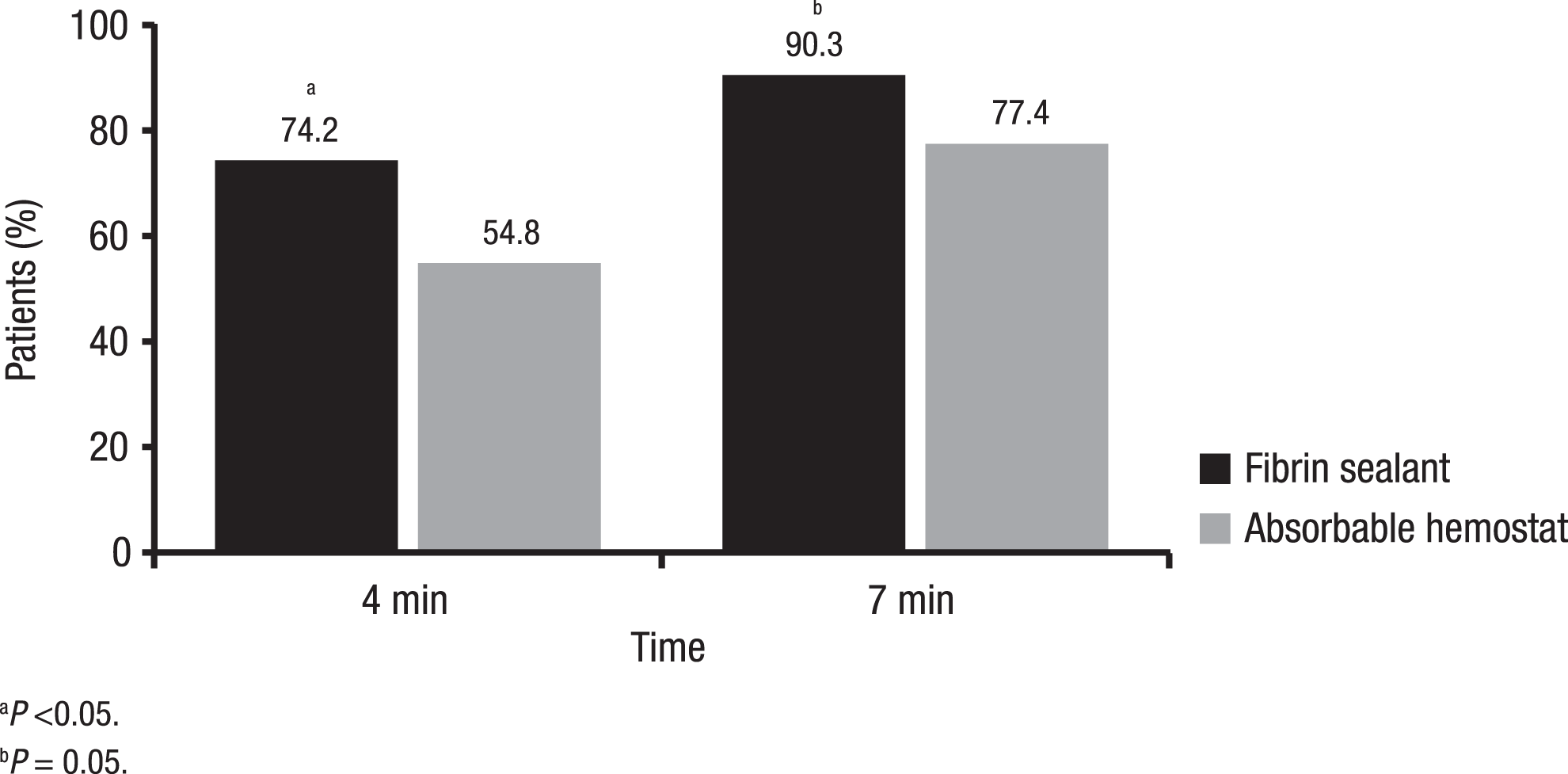
Percentages of patients who achieved hemostasis at 4 and 7 minutes. The percentages of patients in each treatment group who achieved hemostasis at the target bleeding site at 4 and 7 minutes are given.
The overall incidence of treatment failure (included patients who had a TTH >10 minutes or who experienced brisk bleeding during the 10-minute observation period) was significantly lower for patients who received fibrin sealant versus the absorbable hemostat (4.8% vs 17.7%, respectively; P = .0232). No patients who received fibrin sealant experienced brisk bleeding requiring additional hemostatic measures during the 10-minute observation period compared with 3.2% of patients in the absorbable hemostat group (relative risk = 1.03; 95% CI, 0.97-1.12). The incidence of complications that were potentially related to bleeding, including anemia and low hemoglobin levels, was similar between the treatment groups (fibrin sealant, 11.3%; absorbable hemostat, 16.1%; relative risk = 0.70; 95% CI, 0.29-1.67).
A significantly reduced mean TTH was observed for patients randomized to receive fibrin sealant versus absorbable hemostat (2.9 vs 3.7 minutes, respectively; P = .0026). A numerically higher percentage of patients who received fibrin sealant versus absorbable hemostat for the management of mild bleeding achieved hemostasis within 10 minutes (100.0% vs 88.9%, respectively). Similarly, a numerically higher percentage of patients who received fibrin sealant to treat moderate bleeding achieved hemostasis within 10 minutes compared with patients who received absorbable hemostat (87.0% vs 73.1%, respectively).
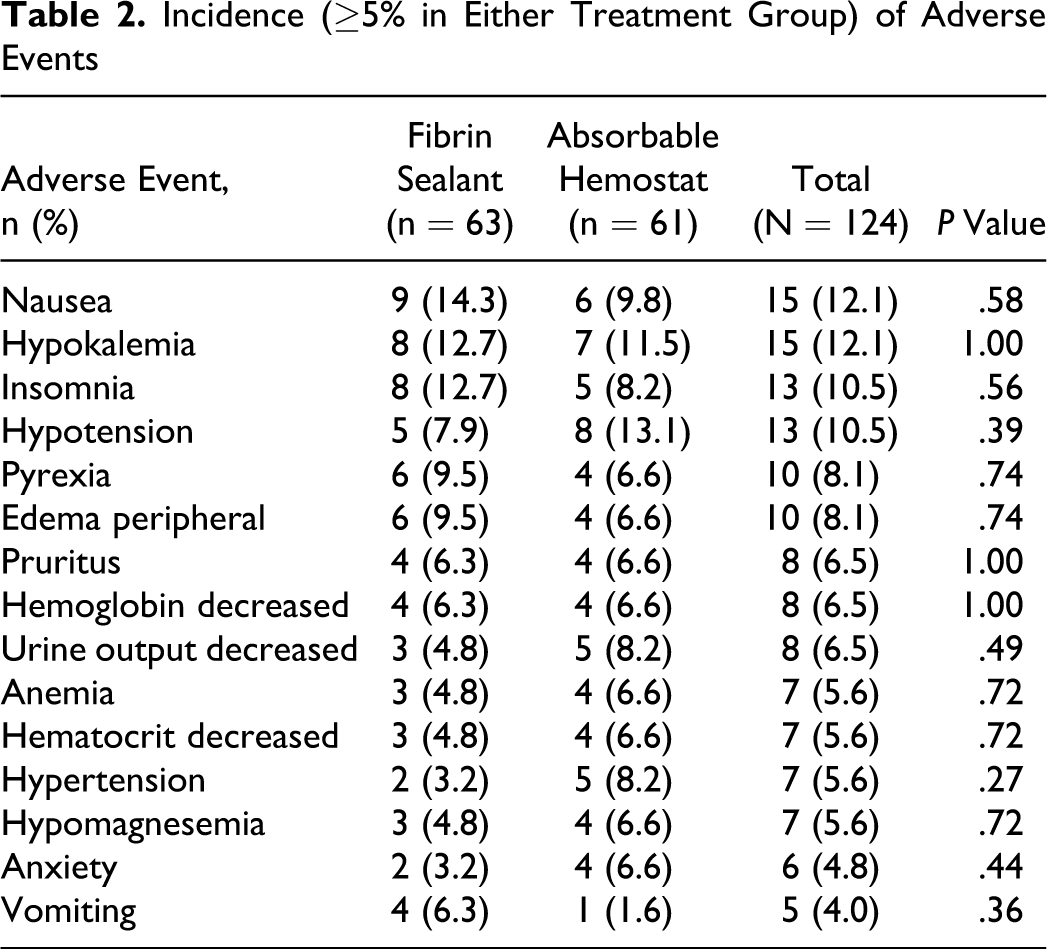
The adverse event profiles were similar between the treatment groups (Table 2 ). The percentages of patients who experienced at least 1 adverse event were similar between the treatment groups (fibrin sealant, 71.4%; absorbable hemostat, 68.9%). No adverse event was judged to be related to either study treatment.
Incidence (≥5% in Either Treatment Group) of Adverse Events
Comment
Fibrin sealants have been shown to be a safe and effective method for achieving hemostasis during a variety of surgical procedures,2–9 and their use during surgery has increased during recent years. Given the widespread use of fibrin sealants and the recent US Food and Drug Administration alert 16 indicating the need for caution in the use of aprotinin, there has been a reappraisal of the need for antifibrinolytic agents in fibrin sealants. Repeated exposure to aprotinin may lead to allergic or potentially fatal anaphylactic reactions,17–19 and tranexamic acid has been linked to alterations in neural tissue growth and adherence. 20 In addition, tranexamic acid-free preparations of fibrin sealant are desirable to allow expanded use in procedures that expose brain and neural tissue.
Results of this randomized, controlled trial demonstrate that the fibrin sealant used in this study is safe and effective as an adjunct to achieve hemostasis for mild-to-moderate bleeding in soft tissue during retroperitoneal or intra-abdominal surgery. The time required to achieve hemostasis was significantly lower for patients who received fibrin sealant compared with those who received absorbable hemostat, and the incidence of adverse events was comparable between the groups.
The use of human-derived biologic agents may incur the risk of transfusion-related illness. This risk is balanced against that of adverse reactions associated with animal-derived components. The biological components of the fibrin sealant used in this study each undergo 2 distinct and separate virus inactivation and/or removal steps during the manufacturing process. 21 Currently available viral inactivation and removal steps, in combination with donor selection and plasma screening processes, help to ensure that manufactured plasma-derived products are pathogen free.22–24 In fact, published data to date of agents used in the preparation of fibrin sealants have not reported any transmission of disease related to the treatment agent.5,25–27
This study may be limited by the fact that surgeons were not blinded to the treatment that was being used. However, a blinded study was not possible given the differences in the application of the fibrin sealant and the absorbable hemostat. Although patients were randomized 1:1 to each treatment group, a numerically higher percentage of patients who had mild bleeding received the fibrin sealant, whereas a numerically higher percentage of patients who had moderate bleeding received the absorbable hemostat; however, this difference in the overall distribution of patients when analyzed by treatment group and severity of bleeding was not statistically significant.
Conclusions
Findings from this study demonstrate that the fibrin sealant used in this trial is safe and effective for achieving hemostasis in soft tissue during elective retroperitoneal or intra-abdominal surgery. The use of this tranexamic acid- and aprotinin-free fibrin sealant should be considered during surgical procedures with mild-to-moderate bleeding when achieving hemostasis quickly is desired and/or necessary.
Footnotes
Acknowledgments
The authors would like to acknowledge the contributions of June Hsu, Rich Kocharian, Gerry Leighton, and David Shah, employees of Ethicon, Inc (Somerville, New Jersey); as well as contributions made by Peter Jones, an employee of Johnson & Johnson Medical Ltd (Livingston, United Kingdom). Craig Fischer has received honoraria from Johnson & Johnson; Christopher Wood served on an advisory board for Ethicon, Inc; and Jessica Shen, Jonathan Batiller, Bob Patel, and James Hart are employed by Ethicon, Inc. This study was supported by OMRIX biopharmaceuticals, Ltd, Kiryat Ono, Israel, and Ethicon, Inc, a Johnson & Johnson company, Somerville, New Jersey, United States. Editorial support for the writing of this manuscript was provided by Ashley O’Dunne, PhD, of MedErgy and was funded by Ethicon, Inc. The authors were not compensated and retained full editorial control over the content of the manuscript. This clinical trial was conducted under the guidance of the following principal investigators at 16 sites: Joel Bauer, MD, Mt. Sinai Hospital, New York, New York; Charles Dunton, MD, Lankenau Hospital, Wynnewood, Pennsylvania; Keith Falcao, MD, St. Agnes HealthCare, Inc, Baltimore, Maryland; Craig Fischer, MD, The Methodist Hospital, Houston, Texas; Henri Ford, MD, Children’s Hospital Los Angeles, Los Angeles, California; Arlan Fuller, MD, Massachusetts General Hospital, Boston, Massachusetts; Barbara Gaines, MD, Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania; Justin Harmon, MD, Cooper University Hospital, Camden, New Jersey; Ronald Lewis, MD, Medical College of Georgia, Augusta, Georgia; Robert Martindale, MD, Oregon Health & Science University, Portland, Oregon; Abed Musa, MD, GYN Oncology Associates, Syracuse, New York; Michael Page, MD, The Iowa Clinic, Des Moines, Iowa; Ruben Ricardo, MD, Miami Research Associates, Miami, Florida; Douglas Swartz, MD and Morteza Yavari, MD, Jacksonville Center for Clinical Research, Jacksonville, Florida; Joseph Trapasso, MD, Lehigh Valley Hospital, Allentown, Pennsylvania; Christopher Wood, MD, The University of Texas M. D. Anderson Cancer Center, Houston, Texas.
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) received no financial support for the research and/or authorship of this article.