
Abstract
We investigated the mechanisms responsible for factor VII (FVII) deficiency in a compound heterozygous Japanese patient with mutations both in the signal peptide and in the catalytic domain. FVII activity (FVII:C) and antigen (FVII:Ag) levels of the patient were 14.5% and 12.5% of those of the normal controls, respectively. In all, 2 heterozygous point mutations were identified in the patient: one was the mutation substituting Pro for Leu-48 in the prepeptide domain of FVII; the other one was a novel mutation substituting Leu for Pro260 in the catalytic domain. FVII activity and FVII:Ag levels in the condition medium that transiently coexpressed the 2 different FVII mutants in baby hamster kidney (BHK) cells were 4.81% and 5.18% of the wild-type FVII. Factor VII defect of the patient may be combined with both impairing endoplasmic reticulum (ER) targeting and altering FVII folding/biosynthesis, but cotransfection of 2 different FVII mutants may interfere with their expression in BHK cells.
Keywords
Human factor VII (FVII) is a vitamin K-dependent glycoprotein that is synthesized in the liver and secreted into the blood. It circulates as a single-chain zymogen of 406 amino acid residues with an apparent molecular weight of 50000. 1 FVII forms a one-to-one stoichiometric complex with tissue factor (TF) in the presence of calcium. Once complexed with TF, FVII is converted to its active form by proteolytic cleavage at a single site (Arg152-Ile153) by various coagulation proteases, including FXa, FIXa, FXIIa, thrombin, and FVIIa. FVIIa is a 2-chain molecule composed of an N-terminal light chain and a C-terminal heavy chain linked by a single disulfide bridge. The light chain largely consists of an N-terminal γ-carboxyglutamic acid-rich domain, followed by 2 epidermal growth factor (EGF)-like domains. The heavy chain contains the serine protease catalytic domain. The TF-FVIIa complex readily activates FX and FIX to ultimately promote fibrin formation.1–3 The gene for FVII is located on chromosome 13 at q34-qter and spans 12.8 kilo base pairs. Exons 1a and 1b encode the 5′ untranslated region and most of the prepro leader sequence, whereas exons 2 to 8 encode the mature protein. 4
Hereditary FVII deficiency is a rare autosomal recessive bleeding disorder, with an estimated prevalence of 1 in 500000 in the general population. 5 Few reports have demonstrated recombinant expression and functional characterization of compound heterozygous mutations in both the prepeptide and the catalytic domain. In the present study, we investigated the mechanisms responsible for FVII deficiency in a compound heterozygous Japanese patient with a Leu-48Pro mutation in the hydrophobic core of the signal peptide and a Pro260Leu mutation in the catalytic domain.
Materials and Methods
Participant
The patient was a 7-year-old Japanese boy with FVII deficiency who was referred for evaluation and coagulation tests after preoperative examination in the otolaryngology department revealed a prolonged prothrombin time. His laboratory values were within normal range except prothrombin time (Table 1 ). He had sometimes experienced epistaxis, that was 0 in a standard bleeding score system. 6 His mother had abnormal bleeding at the time of delivery. Both FVII:C and FVII:Ag levels of the patient were low (14.5% and 12.5% of normal controls). Procoagulation factors apart from FVII were within the normal range, and no FVII inhibitors were found in the plasma. FVII activity and FVII:Ag levels of his father, mother, and a sister were mildly reduced (50% and 50%, 53% and 48%, and 39% and 42%, respectively). FVII activity and FVII:Ag levels of another sister were within the normal range.
Laboratory Results
Collection and Processing of Blood Samples
After obtaining informed consent of the patient’s family members, blood specimens were drawn from the propositus, his parents, and his sisters. Blood samples were collected in 3.2% sodium citrate, and plasma samples were frozen at −80°C until assayed. Genomic DNA was prepared from peripheral blood leukocytes according to previously established methods. 7
Factor VII Assay
FVII activity was assayed using the 1-stage assay with either human recombinant thromboplastin (Dade Innovin, Sysmex, Japan) or immunodepleted FVII-deficient plasma, and analyzed with an Amelung KC4A micro analyzer (Sigma, Germany). The concentrations of FVII:Ag were determined by a modified enzyme-linked immunosorbent assay (ELISA), as previously described. 8 The concentrations of FVII:C and FVII:Ag were expressed as percentages, with 100% defined as the amount present in 1 mL of reference plasma pooled from 40 healthy volunteers.
DNA Isolation and In Vitro Amplification Using Polymerase Chain Reaction
Genomic DNA samples were prepared from peripheral blood leukocytes. The 5′ flanking region contained the promoter region (−420base pair [bp]) and 8 exons, and the adjacent intron junctions. The entire FVII gene of the participant, with the exception of the 1b region, was amplified by PCR using oligonucleotides, as previously described. 9 The promoter region and exon 1a, 2, 3, and 4 were amplified as a single fragment in a reaction mixture containing 500 ng genomic DNA, 0.5 mg of each oligonucleotide, and 1.5 units Taq DNA polymerase (Takara, Otsu, Japan) in 90 mL buffer containing 1.5 mmol/L MgCl2, 10 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, 0.01% gelatin, and 200 mmol/L deoxynucleotide triphosphates (Ultrapure dNTP Set [Pharmacia Biotech, Sweden]). The reaction mixture was amplified using the previously described conditions. 9
Sequencing of Polymerase Chain Reaction Fragments
Polymerase Chain Reaction (PCR) products were electrophoresed on 2% agarose gels in Tris-borate-EDTA buffer (pH 8.3) and stained with ethidium bromide. DNA fragments were then purified from excised agarose gel slices using Gene Clean II (BIO101, La Jolla, California). Purified PCR products were directly sequenced using a Taq Dye Deoxy Terminator Sequencing Kit (Applied Biosystems, Foster City, California) and a 373A DNA sequencer (Applied Biosystems,).
Allele-Specific PCR
Primer-directed allele-specific amplification was used to detect a T→C substitution at nucleotide 38 in exon 1a and a C→T substitution at nucleotide 10696 in exon 8 of the FVII gene that were not detectable by restriction enzyme analysis. To this end, a sense 20-bp oligonucleotide containing the mutant (5′-aggctcctctgccttctgct-3′) or wild-type base (5′aggctcctctgccttctgcc-3′) at its 3′ end were used with a common antisense primer (5′-catgccctcctctaccccat-3′) to amplify a 189-bp fragment from the FVII Leu-48Pro mutant or wild-type allele, respectively. A sense 20-bp oligonucleotide containing the mutant (5′-ctcactgaccatgtggtgcc-3′) or wild-type base (5′-ctcactgaccatgtggtgct-3′) at its 3′ end was used with a common antisense primer (5′-catgccctcctctaccccat-3′) to amplify a 293-bp fragment from the FVII P260L mutant or wild-type allele, respectively. After amplification, 10 μL of the PCR mixture were electrophoresed in a 1.5% agarose gel.
Construction of Expression Vectors
A human FVII cDNA derived from the λHVII565 clone and containing a 66-nucleotide deletion (100-165) with a Hind III/Eco RI linker at each end was prepared as previously described. 10 Site-directed mutagenesis of the FVII coding sequence was performed using the Quick Change Site-Directed Mutagenesis Kit (Stratagene, La Jolla, California) with the following oligonucleotide primer pairs: 5′-tctgccttctgcctgggcttcaggg-3′ and 5′-ccctgaagcccaggcagaaggcaga-3′ for Leu-48Pro, and 5′-accatgtggtgctcctctgcctgcc-3′ and 5′-ggcaggcagaggagcaccacatggt-3′ for Pro260Leu. The oligonucleotide primers were extended by Pfu Turbo DNA polymerase. After thermocycling, the products were treated with Dpn I. Vector DNA containing the desired mutations was then transformed into competent Escherichia coli XL-1 blue cells and grown overnight on LB/ampicillin plates. DNA was prepared from ampicillin-resistant colonies and sequenced to confirm the presence of each mutation. The Hind III-Eco RI fragments of wild-type and mutated FVII were isolated from the FVII cDNA cloned into the pCR vector and ligated into the expression vector pcDNA1.1 (Invitrogen, San Diego, California).
Transient Expression of Recombinant FVII
To investigate the influence of the missense mutations identified in the proband on FVII biosynthesis, we transiently transfected BHK cells with the pcDNA1.1 vector containing either wild-type or mutant FVII cDNAs and compared the levels of secreted FVII in the conditioned media. In addition, we cotransfected a luciferase expression vector and measured luciferase activity in the transfected cell lysates to measure the relative efficiency of transfection.
For transient transfection experiments, BHK cells (ATCC CCL10) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 2 mmol/L
Immunofluorescence Microscopy
Baby hamster kidney cells grown on cover slips (Lab-Tek II chamber slide system [Nunc, Illinois]) were fixed with 4% formaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for 20 minutes at room temperature. After washing with PBS, cells were incubated with anti-human FVII rabbit IgG diluted 1:100 in PBS for 1 hour at room temperature. Cells were washed twice with PBS, then incubated for 30 minutes with fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (1000×) diluted 1:1000 in PBS. After 2 washes with PBS, cells were examined with a fluorescence microscope. 11
Structural Modeling
To examine the environment of the Pro260 residue in the FVII molecule, visualizations using FVII amino acid coordinates extracted from the crystal structure of the FVII zymogen complexed with the inhibitory exosite peptide A-183 (PDB:1JBU) were performed using the graphic software Rasmol version 2.6. All residue numbers correspond to the mature human FVII.
Results
Identification of Mutations in the FVII Gene
The genomic DNA of the patient was analyzed for FVII mutations. Briefly, 2 heterozygous point mutations were identified: a T to C transition at nucleotide 38 in exon 1a, resulting in the substitution of Pro (CCT) for Leu-48 (CTT) in the prepeptide domain of FVII, and a C to T transition at nucleotide 10696 in exon 8, resulting in the substitution of Leu (CTC) for Pro260 (CCC) in the catalytic domain of FVII. No other mutations were identified in the 5′ flanking region containing the promoter region and 8 exons of the FVII gene.
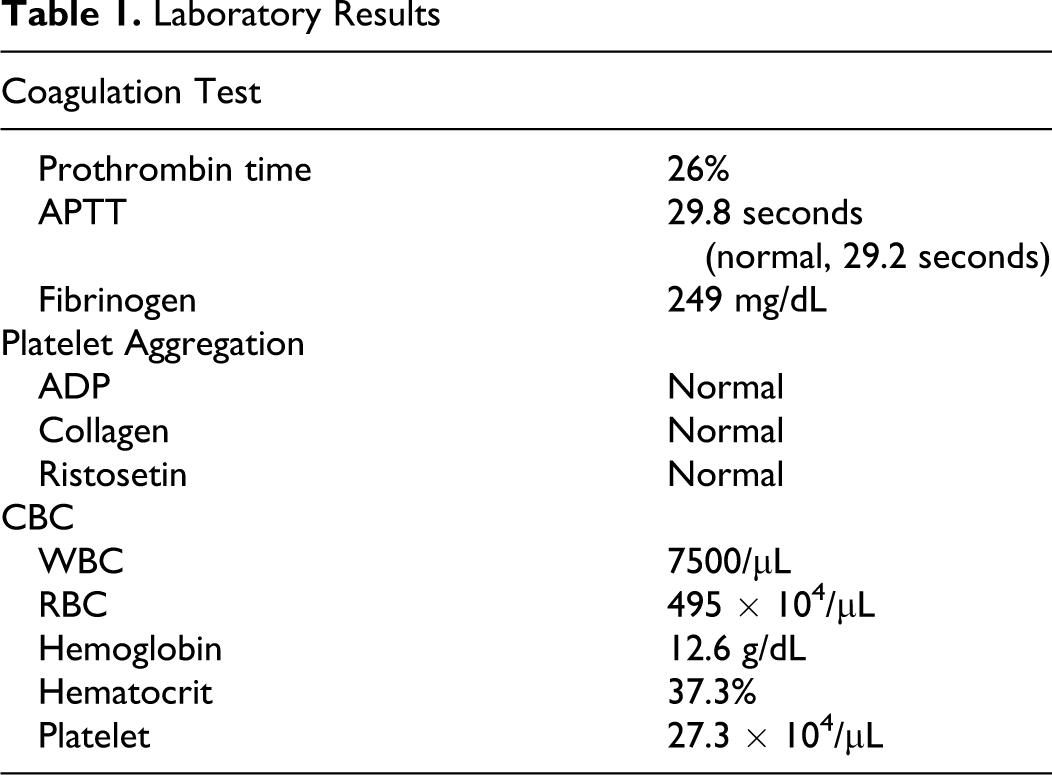
We used allele-specific PCR (ASPCR) to determine the presence of these base substitutions in family members because both the C to T substitution at position 38 and the T to C substitution at position 10696 could not be identified using restriction digest analysis. In the ASPCR for the C to T substitution at position 38, a 189-bp fragment was amplified in all members of the family when using a set of primers designed to specifically amplify the normal allele. However, the use of a set of primers designed to specifically amplify the mutant allele resulted in the amplification of the fragment in the patient, his father and one of the sisters, revealing a heterozygous Leu-48Pro mutation. In the ASPCR for the T to C substitution at position 10696, a 293-bp fragment was amplified in all members of the family using a set of primers designed to specifically amplify the normal allele. Using a set of primers designed to specifically amplify the mutant allele, the fragment was amplified in the patient and his mother, revealing a heterozygous mutation of Pro260Leu (Figure 1 ).
Allele-specific polymerase chain reaction analysis in the patient, his parents, and 2 sisters revealed a C→T substitution at position 38 in the FVII gene that results in Leu-48Pro. (Lower) Using primers for the normal allele (N), fragments of the correct size were amplified in all members of the family. Using primers for the mutant allele (M), fragments of the correct size (189 bp) were amplified from the patient (1), the father (2), and sister 2 (5), but not the mother (3), and sister 1 (4). Allele-specific polymerase chain reaction analysis in the patient, his parents, and 2 sisters detected a T→C substitution at position 10696 in the FVII gene that results in Pro260Leu. (Upper) Using primers for the normal allele (N), fragments of the correct size were amplified in all members of the family. Using primers for the mutant allele (M), fragments of the correct size (293 bp) were amplified from the patient (1) and the mother (3), but not from the father (2), and the 2 sisters (4 and 5).
Transient Expression of Recombinant FVII
FVII activity of mutant FVII (Leu-48Pro) secreted into the conditioned medium was 9.75 ± 1.36% (mean ± SD) of that of the normal recombinant FVII:C, whereas FVII:Ag levels in the conditioned medium were 11.67% ± 1.22% of the normal recombinant protein. On the other hand, FVII:C of mutant FVII (Pro260Leu) secreted into the conditioned medium was 15.30% ± 1.05% of that of the normal recombinant FVII:C, whereas FVII:Ag levels in the conditioned medium were 19.88% ± 4.74% of the normal recombinant protein. Furthermore, to investigate the influence of the 2 different mutations identified in the patient on FVII biosynthesis, cotransfection experiments were performed in BHK cells using a mixture of both FVII Leu-48Pro/pcDNA 1.1 (1 μg) and FVII Pro260Leu/pcDNA 1.1 (1 μg); 2 μg of FVII wild/pcDNA1.1 was used as the control. FVII activity in cells coexpressing the 2 different FVII mutants was 4.81% ± 0.92% of that of the normal recombinant FVII:C in the conditioned medium, whereas FVII:Ag levels were 5.18% ± 1.75% of the normal recombinant protein (Figure 2 ).
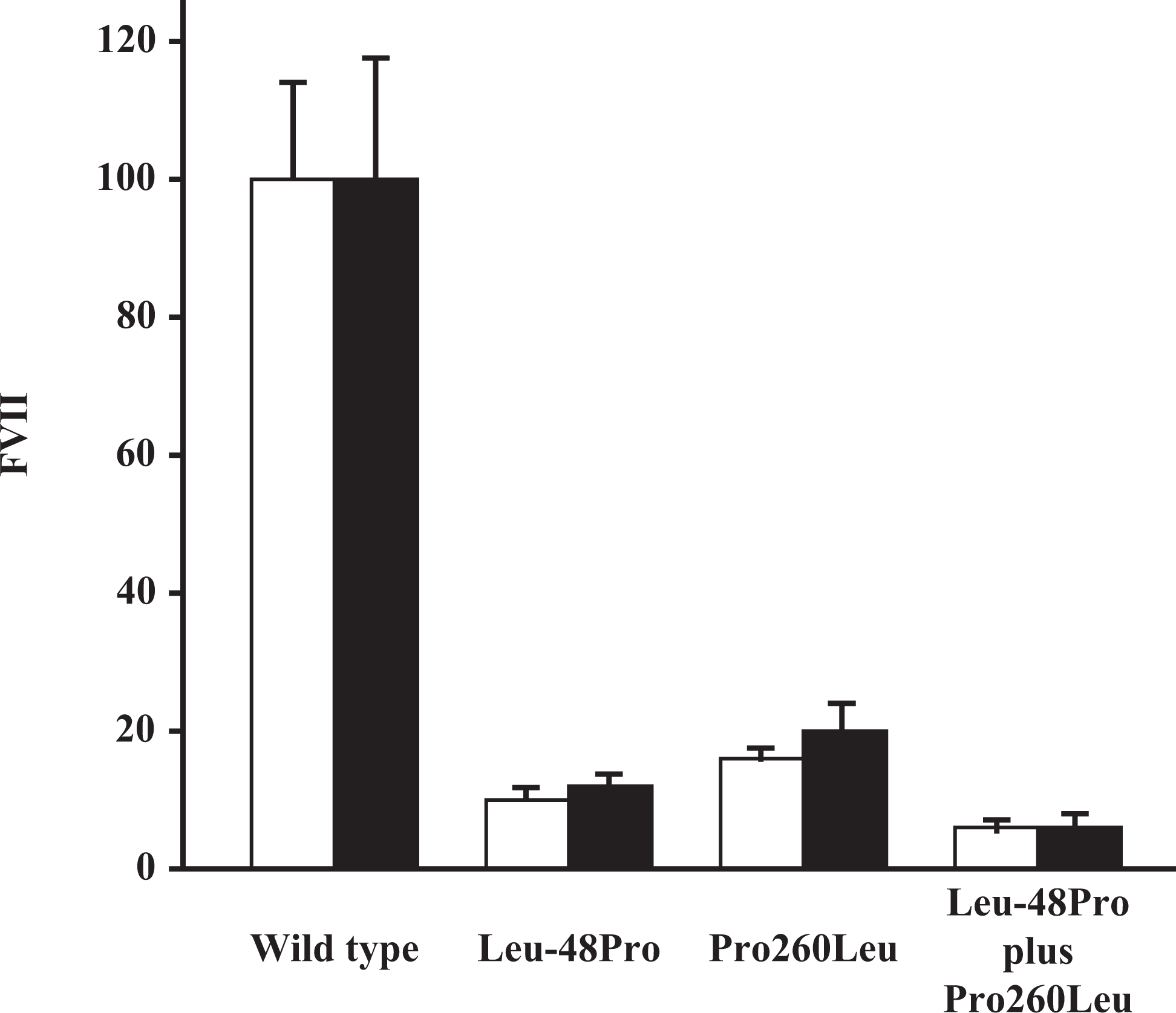
Expression and secretion of wild-type and mutant recombinant FVII in BHK cells. FVII levels in the culture medium and cell extracts harvested from 60-mm dishes 72 hours after transfection with expression vectors were measured by ELISA as described in “Materials and Methods” section. The results are shown as percentage of FVII levels (□, FVII:C; ▪, FVII:Ag), the wild type being defined as 100%.
Using immunohistochemical techniques to detect intracellular FVII:Ag, we found that the 2 FVII mutants were diffusely expressed throughout the cytoplasm in a pattern similar to that of the wild-type FVII (Figure 3 ).
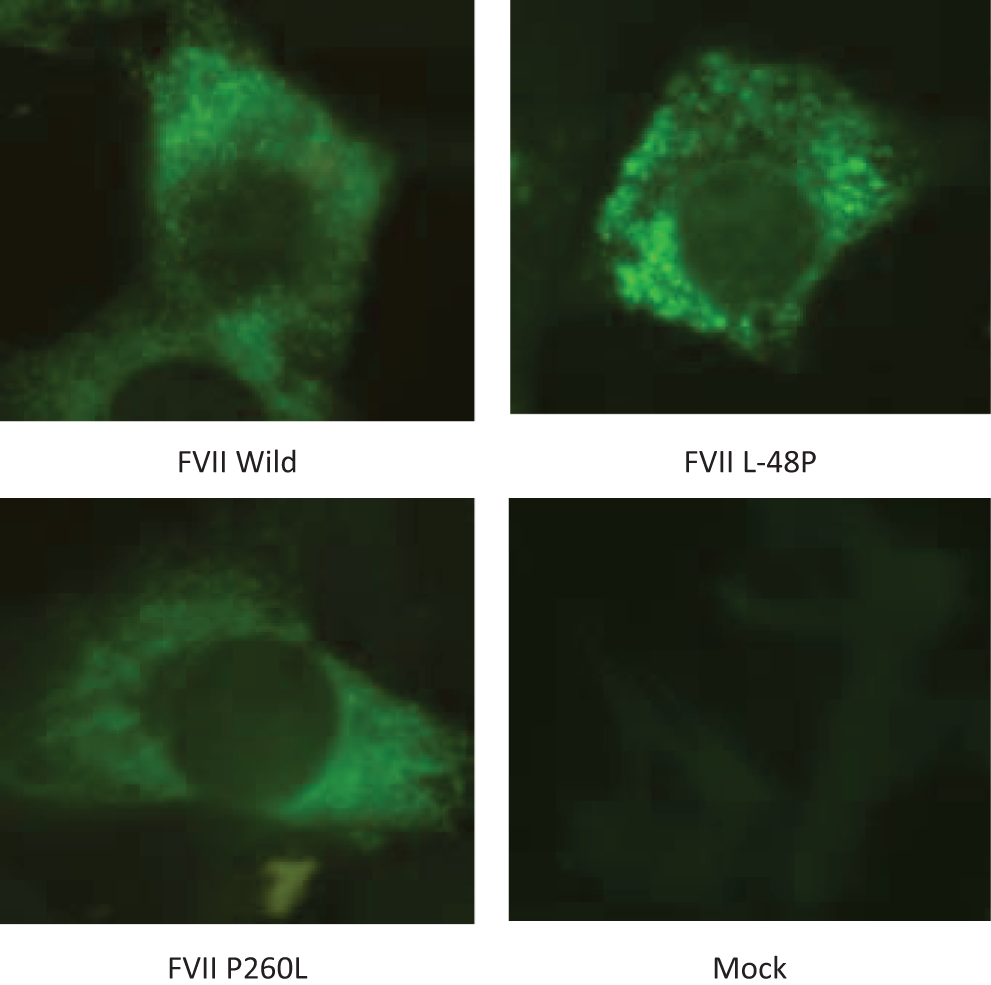
Immunohistochemical localization of wild-type and mutant FVII:Ag in BHK cells. (A) wild-type FVII, (B) FVII Leu-48Pro, (C) FVII Pro260Leu, and (D) Mock.
Discussion
In this study, we investigated a Japanese family with FVII deficiency. The patient was identified as a compound heterozygote, with substitution of Pro for Leu-48 in the prepeptide domain and of Leu for Pro260 in the catalytic domain of FVII. His father and one of his sisters were heterozygous for the Leu-48Pro mutation. His mother was heterozygous for the Pro260Leu mutation (Figure 4 ). At present, more than 170 different naturally occurring mutations in the FVII gene are included in the FVII mutation database. 12 In Japan, 34 different pedigrees with FVII deficiency are included in this database. 13 Of these, 10 cases represent mutations exclusively reported in Japan. The substitution of Leu for Pro260 is a novel mutation associated with FVII deficiency. The homozygous Leu-48Pro mutation, caused by a T to C substitution at nucleotide position 38, was first reported by Ozawa et al. 14 Our results obtained in BHK cells expressing the mutant FVII Leu-48Pro are in agreement with the phenotypic characterization of their patient. It is well known that signal peptides are required for efficient translocation of secretory proteins into the endoplasmic reticulum (ER), and they are cleaved by signal peptidase after the precursor proteins pass across the ER membrane. 15 Secondary structure analysis predicted that the substitution of Pro for Leu-48 reduces the α-helix content of the hydrophobic core in the prepeptide domain of FVII. 16 Ozawa et al described that a slight decrease in hydrophobicity caused by the substitution of Pro for Leu-48 induced inefficient targeting of the protein. 14 Rizzotto et al described that the expression levels of the secreted FVII Leu-48Pro protein were significantly lower than those of wild-type FVII when transiently expressed, and that the markedly reduced FVII levels in cell organelles were the result of impaired ER targeting. 16
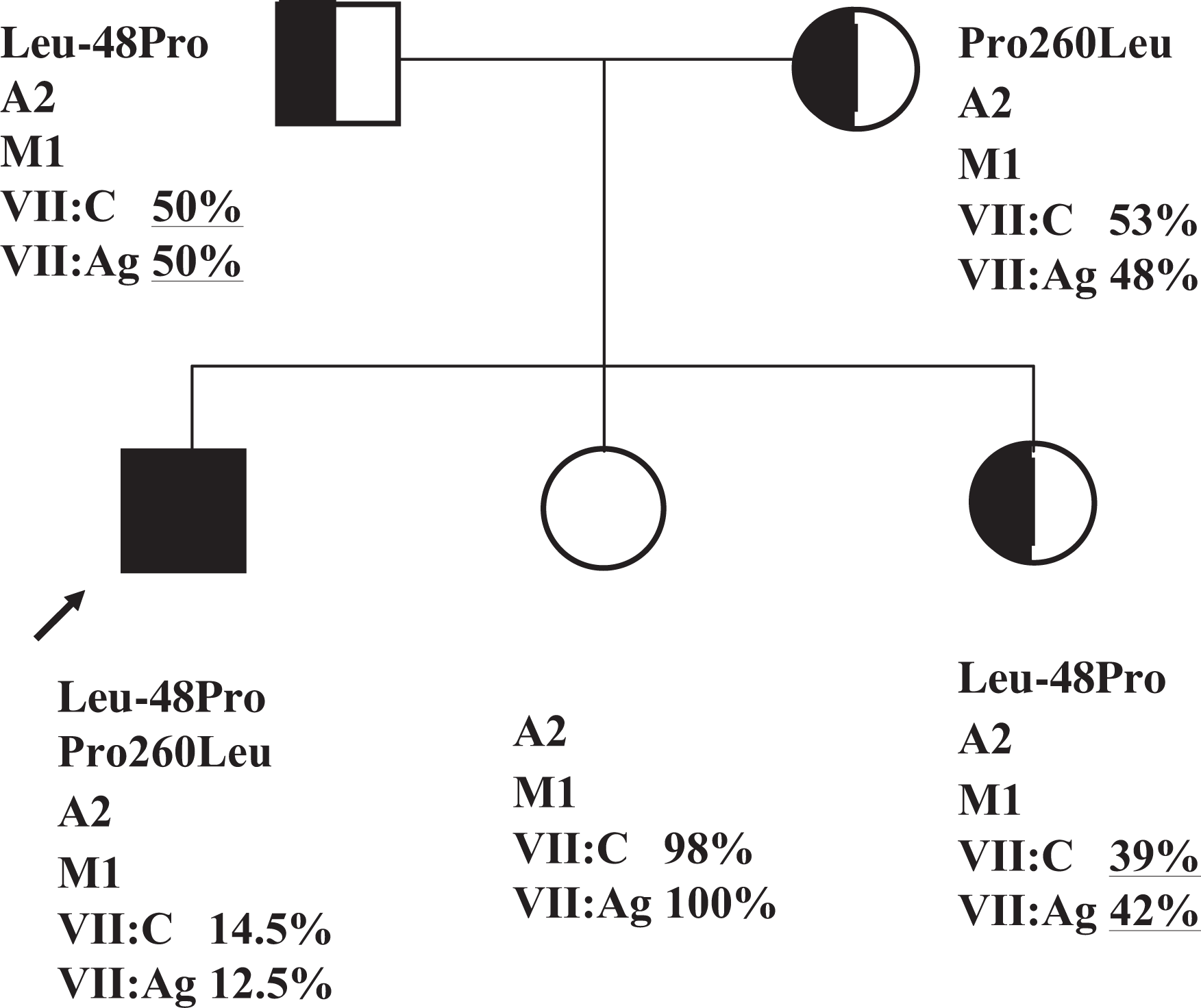
Pedigree of FVII deficiency in compound heterozygotes for mutations in the catalytic domain (Pro260Leu) and the prepeptide domain (Leu-48Pro). The arrow denotes the patient. Compound heterozygous individuals for the Pro260Leu and Leu-48Pro mutations are indicated by a closed symbol, single heterozygotes by half-closed symbols. FVII activity and FVII:Ag levels are expressed as percentages of normal controls. Polymorphism alleles: A1 (insertion) and A2 (no insertion); 10-bp insertion at nucleotide −323, (M1) and (M2); Arg353Gln.
Molecular modeling of FVII (PDB:1JBU) revealed that Pro260 is located on the outside of the 3D structure of the FVII heavy chain. Pro260 is a highly conserved residue in all vitamin K-dependent coagulation factors and within rat, rabbit, bovine, mouse, and human FVII. The structural properties of Pro give it an exceptional conformational rigidity as compared to other amino acids. On the other hand, Leu is a hydrophobic amino acid with a branched hydrocarbon side chain and is generally buried in folded proteins. Its substitution might alter FVII folding/biosynthesis. An analogous Pro287Leu substitution in the FIX protein has been reported in patients with CRM-negative hemophilia B. 17
Coexpression of the 2 different FVII mutants (FVII Leu-48Pro and FVII Pro260Leu) resulted in lower levels of secreted FVII than when either mutant is expressed alone. If mutant FVII Leu-48Pro and FVII Pro260Leu would be synthesized in equal amounts in BHK cells, the FVII:C of the compound heterozygous patient could be mimicked. However, the FVII:C in the coexpression experiment in BHK cells was lower than in the proband. Cotransfection of the 2 different FVII mutants may affect each other’s expression in BHK cells.
Fromovich-Amit et al described that coexpression of Ala244Val and Arg353Gln, but not of Ala294Val and Arg353Gln, had an additive effect on FVII secretion in BHK cells. 18 Kravtso et al reported that the levels of secreted protein are significantly reduced in human embryonic kidney 293 fibroblasts, expressing either FXI Gly350Glu, FXI Gly400Glu, or FXI Tyr569Ser, and that coexpression of FXI Gly400Glu and FXI Tyr569Ser, but not FXI Gly350Glu, with wild-type FXI reduces wild-type secretion by about 50%. 19
Differences between the laboratory phenotype of FVII deficiency caused by compound heterozygous mutations in both Leu-48Pro and Pro260Leu and the results of coexpression of the 2 mutant proteins remain to be clarified.
We identified a compound heterozygous patient for point mutation of Leu-48Pro in the prepeptide domain and of Pro260Leu in the catalytic domain of FVII. Coexpression experiments demonstrated that the FVII deficiency of the patient was caused by compound heterozygous mutations of both Pro260Leu and Leu-48Pro.
On the other hand, the clinical correlations of the molecular findings are unclear. In general, 71% of homozygous and 50% compound heterozygous participants were symptomatic. The clinical manifestations of the homozygous participants were characterized by epistaxis, gum bleeding, easy bruising, and so on. The pattern of bleeding symptoms among compound heterozygous patients was similar to that of the homozygous patients. 20 However, the homozygous with substitution of Pro for Leu-48 had no hemorrhagic episodes. 14 Genotype-phenotype relationships indicate the presence of major environmental and/or extragenic components modulating expressivity of FVII deficiency. 21
Footnotes
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) received no financial support for the research and/or authorship of this article.