
Abstract
Introduction
Aprocitentan, is an orally active, once-daily administered, dual endothelin receptor antagonist that was recently approved for the treatment of hypertension in combination with other antihypertensive drugs, to lower blood pressure in adult patients who are not adequately controlled on other drugs.
Methods
The potential of aprocitentan to affect the QT interval was investigated in this thorough QT/QTc study that was performed as a multiple-dose, randomized, double-blind, placebo- and moxifloxacin-controlled (open-label), crossover study in healthy male and female subjects. Concentration-QT modeling, using a linear mixed-effects model, was performed on data extracted from continuous Holter electrocardiogram recordings.
Results
Of the 48 subjects enrolled in the study, 32 subjects completed study treatment. The main reasons for stopping study treatment were the occurrence of adverse events and withdrawal of consent. The dosing regimen of 25 mg aprocitentan o.d. (the highest approved therapeutic dose in the European Union; Cmax = 3.68 µg/mL) was safe and well tolerated; the supratherapeutic dosing regimen of 100 mg aprocitentan o.d. showed limited tolerability. Results indicated that ΔΔQTcF increased with increasing aprocitentan concentrations. The predicted upper bound of the 2-sided 90% ΔΔQTcF confidence interval exceeded the 10 ms threshold of regulatory concern at 16.10 µg/mL, which was close to the geometric mean maximum concentration (ie, 16.77 µg/mL) obtained with a 100 mg supratherapeutic dose.
Conclusions
At the highest therapeutic dose of 25 mg, there was no clinically significant prolongation of QTc. The risk of QT prolongation with therapeutic doses of aprocitentan is considered low.
Introduction
High blood pressure (BP), also known as hypertension, is one of the most important risk factors for cardiovascular disease and, globally, is a cause for increased morbidity and mortality.1,2 One subset of patients that do not achieve control of their BP are known as resistant hypertensives.
Resistant hypertension (RHT) is defined as an elevation in BP that remains above a patient's individualized goal despite the concurrent use of 3 antihypertensive agents of different classes. 3 These commonly include a long-acting calcium channel blocker, a blocker of the renin-angiotensin system, and a diuretic, all administered at maximum or maximum tolerated doses and at the appropriate dosing frequency. 3 Before diagnosis, factors that could preclude RHT should be ruled out; the most important being the use of substandard BP measurement techniques, nonadherence to prescribed medication, and the white coat effect. 4 For these reasons, the prevalence of RHT is difficult to discern and has been reported in the range between 9% and 18%. 5 Patients with RHT often are of older age and have comorbidities such as diabetes mellitus, chronic kidney disease, and obstructive sleep apnea and are at higher risk of complications including cardiovascular disease, stroke, kidney failure, and death.6–9 Compared to normotensive subjects or patients with hypertension controlled with ≤2 antihypertensive medications, patients with RHT have often lower plasma renin activity, higher aldosterone, and brain natriuretic peptide levels, and a salt-sensitive form of hypertension. 10 Resistant hypertension patients generally respond well to aldosterone receptor blockade though this comes with several safety concerns such as hyperkalemia and renal insufficiency.11,12 Such characteristics of RHT may call for treatments that work through different pathways, exhibiting different mechanisms of action, one of them being the endothelin (ET) system. Since its discovery in 1988, the physiological and pathophysiological roles of endothelin-1 (ET-1), one of the most potent vasoconstrictor peptides known, have been intensively researched.13,14 Endothelin-1 is part of the ET family that also comprises ET-2 and ET-3. Endothelin-1 is the most abundant peptide that besides its crucial role in regulating BP contributes to almost all aspects of physiology and cell function via its 2 receptors ETA and ETB.14–18 In low-renin models of hypertension, an increase in ET-1 is observed and plasma renin activity and ET-1 are inversely correlated in hypertensive patients.19,20 It is therefore reasonable to consider that insufficient control of BP can be, in part, attributed to an incomplete coverage of all underlying pathological pathways. Given the current limitations in treatment options in RHT the application of agents acting through a different mechanism, such as ET-1 receptor antagonism (ERA), can therefore be a valuable addition to the armamentarium in achieving BP control. Indeed, an ETA selective ERA, darusentan, showed a significant decrease in BP in patients with RHT in a phase 3 trial but could not replicate findings in a second phase 3 trial. Moreover, its development was discontinued after the occurrence of serious adverse cardiac events. 21
Aprocitentan, an orally active dual ERA, has emerged as a potential new treatment for RHT. Aprocitentan has a half-life of approximately 44 h and a low drug–drug interaction potential.22–25 Based on its pharmacokinetic (PK) profile, no dose adjustments are needed for age, sex, race/ethnicity, mild/moderate hepatic impairment, and any degree of renal impairment.25–28
In preclinical and clinical settings aprocitentan demonstrated to efficiently lower BP.29–31 Most notably, in the recently completed multicenter, blinded, randomized, parallel-group phase 3 study (PRECISION) doses of 12.5 and 25 mg aprocitentan showed a significant decrease in systolic and diastolic BP in subjects that had documented uncontrolled hypertension in spite of the use of 3 or more antihypertensive agents. 29 The most common adverse events (AEs) observed were edema or fluid retention that is typically reported for any ERA. 29
The favorable benefit–risk profile of aprocitentan resulted in the approval by the FDA for a daily dosing regimen of 12.5 mg in March 2024 and approval by the European Commission for 12.5 mg with the possibility to increase to 25 mg in July 2024.32,33
As part of the clinical development of aprocitentan, its potential to delay cardiac repolarization was investigated.34,35 An early investigation had already taken place as part of the First-into-Human (FIH) study of aprocitentan, which indicated no apparent drug-induced electrocardiogram (ECG) effects based on time-matched analysis of QTc and no QTc prolongations up to plasma levels of 10 µg/mL. 25 At that stage of development, the number of subjects included in the investigation of QTc prolongation was relatively low, with no possibility of comparison with the positive control moxifloxacin. Therefore, the current thorough QT (TQT) study was designed to provide further evidence that aprocitentan at therapeutic concentrations does not prolong QTc.
Methods
Study Design
The study followed the principles of the Declaration of Helsinki and good clinical practice. The United States Food and Drug Administration (US FDA) Interdisciplinary Review Team was consulted and agreed to the study design. An independent ethics committee (Ethics Committee of the Medical Council of Baden-Württemberg, Stuttgart, Germany) reviewed and approved the study protocol and its amendments as well as the national health authority of Germany (Federal Institute for Drugs and Medical Devices of Germany - BfArM), and the study was executed at CRS Clinical Research Services Mannheim GmbH, Mannheim, Germany.
This study was conducted in a prospective, single-center, randomized, double-blind, placebo- and moxifloxacin-controlled (open-label), 4-way crossover phase 1 design (NCT04281342) in 48 healthy male and female subjects who provided written informed consent to the study prior to any study procedure.
Following a screening evaluation, eligible subjects underwent 4 periods in which subjects were to receive in randomly assigned sequence, using a Willams square design, each of the following 4 treatments:
Treatment A—aprocitentan highest used therapeutic dose level. After the check-in on day 1, subjects received repeated oral doses of 25 mg aprocitentan o.d. from day 1 to day 10 as a single tablet and 3 tablets of aprocitentan-matched placebo, followed by 18 days of observation. Treatment B—aprocitentan supratherapeutic dose level. After check-in on day 1, subjects received repeated oral doses of 100 mg aprocitentan o.d. from day 1 to day 10 as 4 tablets of 25 mg aprocitentan, followed by 18 days of observation. Treatment C—placebo. After check-in on day 1, subjects received repeated oral doses of 4 tablets of aprocitentan-matching placebo o.d. from day 1 to day 10, followed by 18 days of observation. Treatment D—moxifloxacin. After check-in on day 1, subjects received repeated oral doses of 4 tablets of aprocitentan-matching placebo o.d. from day 1 to day 9. On day 10, they received a single oral dose of 400 mg moxifloxacin, followed by 18 days of observation.
The 4 periods were separated by a minimum of 2 days. If there were more than 7 days between periods, additional laboratory tests were performed to confirm that subjects were still eligible to receive treatment. An end-of-study examination took place 20 to 25 days after the last study treatment administration in Period 4 or, in the event of early discontinuation, after the last study treatment intake in any period. A final follow-up for safety was performed 30 to 33 days after the last study treatment intake.
The therapeutic dose of 25 mg aprocitentan corresponded to the highest dose tested in the PRECISION phase 3 study as well as the highest therapeutic dose that can be used in clinic. 29 The supratherapeutic dose of 100 mg aprocitentan, that was well tolerated in the FIH study, was predicted to cover 4 and 2 times the exposure of a mean population and a high exposure scenario for subjects using 25 mg aprocitentan, respectively. 25
Study Population
A total of 30 evaluable subjects were needed to allow sufficient power to exclude a ΔΔQTcF (placebo-corrected change from baseline QT interval corrected for heart rate with Fridericia's formula) in subjects treated with aprocitentan exceeding 10 ms. The study was set up to detect a ΔΔQTcF of 10 ms with a power of 95%, assuming an SD of 8 ms. The calculations were based on 10 000 simulations of the study setup with an assumed QT effect of 3 ms at maximum plasma concentration (Cmax), using the one-sided Student t test for paired observations at the 5% significance level. 36
Initially, it was planned to recruit 36 subjects to achieve 30 evaluable subjects. As the study was conducted during the Coronavirus Disease (COVID)-19 pandemic, a higher than anticipated number of subjects did not complete all study treatments. Therefore, 48 healthy male and female subjects between 19 and 55 years were included in this study. Good health was assessed based on medical history, physical examination, 12-lead ECG, and routine clinical laboratory tests. Specifically, subjects were eligible if QTcF < 450 ms for male subjects and <470 ms for female subjects, QRS < 110 ms, PR < 220 ms, and resting HR > 50 b/min and <90 b/min and the 12-lead ECG did not show any clinically relevant abnormalities. Further, subjects could not participate if they had personal or family history or presence of rhythm disorders or hypokalemia.
Women of childbearing potential were required to have negative pregnancy tests at screening and on day 1 and had to use a reliable method of contraception from screening until 30 days after last study treatment intake. As the subjects were healthy, they were not allowed to take any concomitant medication except for contraceptives, hormone replacement therapy, and medications for treatment of AEs.
As this study was conducted during the COVID-19 pandemic, subjects had to adhere to COVID-19 precautions and guidance as per country regulation as well as undergo regular testing.
Study Treatment
Aprocitentan was provided as film-coated tablets containing 25 mg aprocitentan. Aprocitentan-matching placebo was provided as film-coated tablets that matched in size, weight, and appearance, formulated with the same excipients as the aprocitentan tablet. Moxifloxacin was commercially acquired as film-coated tablets of 400 mg moxifloxacin (moxifloxacin-ratiopharm®).
Study treatment was dispensed by a nonblinded pharmacist at the study site according to a randomization list presenting the different sequences per subject generated using SAS version 9.4. The list was provided prior to first dosing of the first subject. The randomization codes were kept strictly confidential and were accessible only to authorized people not involved in the conduct and analysis of the study until unblinding.
Eligible subjects received their randomization number on day 1 of the first period prior to start of study procedures/assessments. To maintain the blind during day 1 to day 10, placebo tablets were given alone or together with the study treatment to match the number of tablets given per day. The only exception was for day 10 in Treatment D when a single moxifloxacin tablet was administered open label.
Pharmacokinetic Blood Sampling
Blood samples (approximately 1.2 mL) were collected in each treatment with full profiles taken on day 1 and 10 (predose and 1, 2, 3, 4, 6, 7, 8, 9, 10, 12, 15 h postdose) and trough concentrations on days 2 to 9 as well as 24 h postdose after last dose on day 10 (ie, day 11). Plasma concentrations of aprocitentan were determined using a validated liquid chromatography method coupled to a tandem mass spectrometer, as previously described. 25 In the present study, the lower limit of quantification (LOQ) was 5 ng/mL, the interbatch precision was ≤6.0%, and the interbatch accuracy was in the range from −0.5 to −1.6%.
There appeared no need to measure moxifloxacin concentrations as the evaluation of the ECG data unequivocally demonstrated assay sensitivity.
12-Lead Continuous Holter Monitoring and Extraction of ECG Intervals
Continuous 12-lead ECGs were recorded over at least 24 h on days 1 to 2 and days 10 to 11 using digital Holter recorders (CM 3000-12, GETEMED AG) which were supplied and supported by the ECG core laboratory (Nabios GmbH). Of these recordings triplicate ECGs were extracted at predetermined time points for day 1 (−1, −0.75, −0.5 h predose and 1, 2, 3, 4, 8, 12 h postdose) and day 10 (−0.5 h predose and 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 15, 24 h postdose). Extraction of the ECGs and subsequent determination of the ECG intervals (QTcF, HR, PR, and QRS) were performed by trained study staff of the ECG core lab that were blinded to treatment. In addition, one ECG for each time point was assessed by a board-certified cardiologist. Besides an overall assessment, qualitative ECG findings regarding rhythm, conduction, morphological abnormalities, presence of myocardial infarction, ST-segment deviations, T-wave morphology, and presence of U-waves were recorded, if present.
Safety Evaluations
Tolerability and safety were evaluated descriptively by recording of treatment-emergent AEs (TEAEs), serious AEs (SAEs), clinical laboratory variables, vital signs, and 12-lead ECG predose and at predetermined time points during each treatment. Treatment-emergent AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA version 23.0).
Data Analysis and Statistical Methods
Four analysis sets were used for evaluation:
The All-treated set (ATS) was used for evaluation of safety and tolerability and included all subjects who received at least one dose of study treatment. The PK set used for evaluation of PK included all subjects in the ATS who had at least one valid concentration of aprocitentan. In addition, subjects could not deviate from the protocol that could have affected the PK evaluation. The ECG set used for evaluation of assay sensitivity of moxifloxacin included all subjects in the ATS with measurements at baseline as well as on-treatment with at least one postdose time point with a valid ΔQTcF value. The PK/ECG set used for evaluation of the cardiodynamic ECG endpoints included all subjects in the PK and ECG set with at least one time-matched postdose PK and QTcF pair of measurements, and subjects on placebo in the ECG set.
Tolerability and safety parameters were analyzed descriptively.
The plasma PK parameters were derived by noncompartmental analysis (Phoenix version 8.0, Certara) based on the actual blood sample time points. The measured individual plasma concentrations of aprocitentan on day 1 and day 10 were used to directly obtain Cmax and time to Cmax (tmax). Values below LOQ were set to zero. The area under the plasma concentration-time curve during one dosing interval (AUCτ) on these days was calculated according to the linear trapezoidal rule using the measured concentration-time values above the LOQ. The accumulation index (AI) was defined as the AUCτ on day 10 / AUCτ on day 1. It was assumed that Cmax and AUCτ were log-normally distributed. Pharmacokinetic parameters were summarized using geometric means and 2-sided 95% confidence intervals (CIs) or median and range for tmax.
Assay sensitivity (ie, the ability to detect a QT prolongation in the study if present) was investigated using the moxifloxacin Holter data. A one-sided one-sample Student t test was applied to test if the mean placebo-adjusted ΔQTcF (ΔΔQTcF) was greater than 5 ms with α = 0.05/3. Assay sensitivity was met if the null hypothesis was rejected for at least one time point on day 10 at 2, 3, or 4 h after moxifloxacin dosing using the Bonferroni correction for multiple testing.
The primary analysis was based on concentration-QTcF (c-QTc) modeling by Garnett et al. 37 Here, the relationship between aprocitentan plasma concentration and QTcF was investigated with the intent of excluding an effect of population average ΔΔQTcF > 10 ms at clinically relevant plasma concentrations. A linear mixed-effects model was employed38,39 in which ΔQTcF was related to the fixed effects: intercept and slope of the c-QTc relationship, study treatment, QTc baseline centered by QTc baseline average, and time, as well as the random effects: subject-specific intercept and slope.
A by-time-point analysis was conducted investigating ΔΔQTcF, ΔΔHR, ΔΔPR, and ΔΔQRS at each assessment after dosing. A linear mixed-effects model was developed in which time point, treatment, period, sequence, interaction between treatment and time point, and centered baseline were fixed effects. Interindividual variability was included on intercept, assuming a normal distribution. An unstructured covariance matrix was specified, and an additive residual error term was used. Other evaluations were conducted using graphical displays and descriptive summaries. Categorical outliers were summarized in frequency tables and were defined as follows: absolute QTcFOBS values >450 and ≤480 ms, >480 and ≤500 ms, or >500 ms; increases in QTcFOBS from the observed individual baseline of each treatment of >30 and ≤60 ms, or >60 ms; increase in PROBS from the observed individual baseline of each treatment >25% resulting in PROBS > 200 ms; increase in QRSOBS from the observed individual baseline of each treatment > 25% resulting in QRSOBS > 120 ms; decrease in HROBS from the observed individual baseline of each treatment >25% resulting in HROBS < 50 bpm ; increase in HROBS from the observed individual baseline of each treatment >25% resulting in HROBS > 100 bpm . For each of these categories, a categorial outlier was deemed present if observed in all replicates at the time point. Treatment-emergent abnormalities in T-wave morphology and abnormal U-wave presence were listed and tabulated.
Results
Subject Demographics and Disposition
In total, 48 subjects were included in the study and received at least one dose of study treatment. Of these, 32 subjects completed all 4 treatments and 16 discontinued the study treatment. The main reasons for discontinuation were the occurrence of AEs (7 subjects) and withdrawal of consent (4 subjects), with other reasons only given for 1 or 2 subjects. The number of subjects available for evaluation of PK, ECG, PK/ECG, and safety and tolerability were 44, 48, 39, and 48, respectively.
Demographic characteristics were similar across analysis sets. There were 29 male and 19 female healthy subjects with a mean age of 41.5 years and mean body mass index of 26.6 kg/m2. All except for 2 subjects were White/Caucasian. Except for 7 females that used hormonal contraceptives and 1 female using hormone replacement therapy, none of the subjects took previous/concomitant treatments.
Pharmacokinetics
Mean plasma concentration versus time profiles are shown in Figure 1, and a summary of the aprocitentan plasma PK parameters is presented in Table 1. For both aprocitentan doses, peak plasma concentrations were reached on days 1 and 10 at a median of 6 and 4 h, respectively. Steady-state conditions were reached by day 7 (data not shown). Cmax and AUCτ increased approximately dose-proportionally with an accumulation at steady-state of approximately 3.

Schematic Study Design.
Summary of Plasma PK Parameters of Aprocitentan Assessed on Day 1 and Day 10.a

Abbreviations: AI, accumulation index; AUCτ, area under the plasma concentration-time curve during one dosing interval; CI, confidence interval; Cmax, maximum plasma concentration; N, number of subjects with available value; tmax, time to reach maximum plasma concentration.
Data expressed as geometric mean (95% CI) or as median (range) for tmax.
Cardiodynamic ECG Analysis
Assay sensitivity
The ability to detect a QT effect was demonstrated using the Holter data recorded after administration of 400 mg moxifloxacin. The null hypothesis was rejected for all 3 time points 2, 3, and 4 h after moxifloxacin administration on day 10 (P < 0.05/3 = 0.017) (Table 2).
Mean Observed ΔΔ QTcF (ms) Measured Predose and After Administration of a Single Dose of 400 mg Moxifloxacin on Day 10.
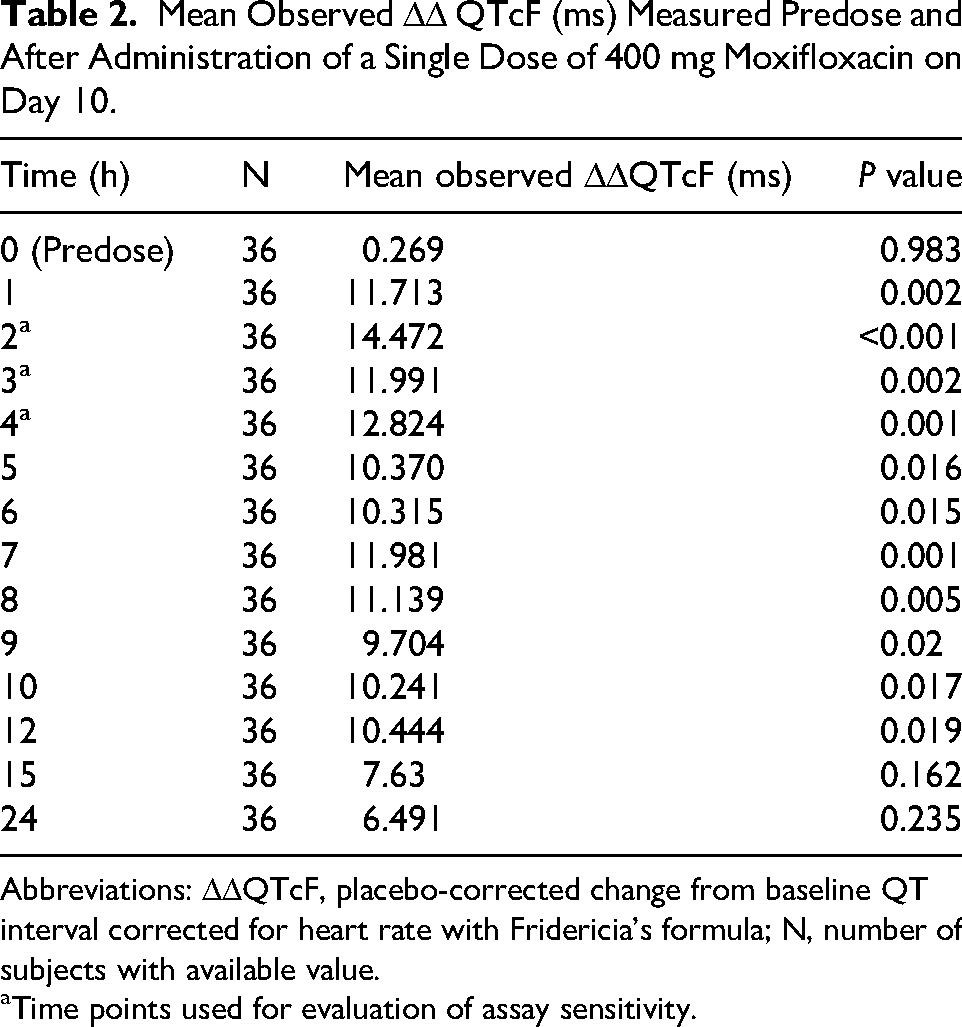
Abbreviations: ΔΔQTcF, placebo-corrected change from baseline QT interval corrected for heart rate with Fridericia's formula; N, number of subjects with available value.
Time points used for evaluation of assay sensitivity.
Primary c-QTc analysis
For the c-QTc analysis a linear mixed-effects model was developed, relating ΔQTcF to aprocitentan plasma concentrations. The slope was estimated at 0.354 ms.mL/µg and was significantly different from 0 ms.mL/µg (P <0 .001). The intercept of −1.557 ms was not significantly different from 0 ms (P = −0.402), while the effect of treatment on the intercept (2.543 ms) was significantly different from 0 (P < 0.001). This resulted in an intercept for subjects on active treatment of 0.986 ms. The slope and intercept were negatively correlated, indicating that subjects with a greater slope were estimated to have a smaller intercept. Using this c-QTc relationship, an increase in ΔΔQTcF with increasing concentrations of aprocitentan could be observed as shown in Figure 2. At the geometric mean steady-state Cmax of 25 mg (highest therapeutic dose; 3.68 µg/mL) and 100 mg (supratherapeutic dose; 16.77 µg/mL), the effect on ΔΔQTcF was 3.846 (3.267-4.426) and 8.482 (6.554-10.411) ms, respectively. The predicted 2-sided 90% CI exceeded the 10 ms threshold at a concentration of 16.10 µg/mL aprocitentan.

Arithmetic (± SD) Plasma Concentration Versus Time Profiles of 25 (Highest Therapeutic) and 100 (Supratherapeutic) mg Aprocitentan Administration on Day 1 (Left Panel) and Day 10 (Right Panel) (N = 44).
By-time-point analysis
Using a linear mixed-effects model, ΔΔQTcF is visualized over time in Figure 3. After treatment with both 25 and 100 mg aprocitentan, ΔΔQTcF increased over time. The 90% CI crossed the threshold of regulatory concern of 10 ms following multiple daily doses of 100 mg aprocitentan. Visualizations over time of ΔΔHR, ΔΔPR, and ΔΔQRS showed a not clinically significant increase in ΔΔHR (<10 b/min) while there were no changes in PR and QRS intervals.

Concentration-QTc (c-QTc) Analysis: Mean ΔΔQTcF Versus Concentration. Red Horizontal Line, 10 ms Threshold of Regulatory Concern; Black Line and Height of Shaded Area, Mean Model-Predicted ΔΔQTcF with 2-Sided 90% Confidence Interval; Width of Blue Highlighted Shaded Areas, 2-Sided 95% Confidence Interval of Geometric Mean Maximum Concentration per Dose Group. Abbreviations: o.d., Once Daily; ΔΔQTcF, Model-Predicted Placebo-Corrected Change from Baseline QT Interval Corrected for Heart Rate with Fridericia's Formula.

By-Time-Point Analysis: Mean ΔΔQtcf Versus Time by Treatment. Error Bars, 2-Sided 90% Confidence Intervals. Abbreviations: BTP, by Time Point; o.d., Once Daily; ΔΔQtcF, Placebo-Corrected Change from Baseline QT Interval Corrected for Heart Rate with Fridericia's Formula.
Categorical outliers
Few categorical outliers were identified. There were no QTcF values of 480 to 500, >500, or ΔQTC >60 ms observed in this study, while other categorical outliers were seen in subjects treated with moxifloxacin or for whom the observation had been present already at baseline. There were no outliers with a significant decrease in HR; one subject had an increase of 25% resulting in HR >100 bpm following treatment with 100 mg aprocitentan. No outliers in PR or QRS were identified and no treatment-emergent U-waves were present. Treatment-emergent T-wave abnormalities of no clinical significance were reported in few subjects, mostly after treatment with moxifloxacin.
Safety and Tolerability
No SAEs were reported. Seven subjects discontinued the study treatment due to an AE (ie, irrespective of causality to treatment); 4 discontinued after treatment with 100 mg aprocitentan and 3 after treatment with placebo. All AEs resolved by the end of the safety follow-up period and were of mild to moderate intensity.
Most TEAEs were reported after treatment with 100 mg aprocitentan (79.1% of the subjects) when compared to other treatments (45.2%, 48.8%, and 39.0% of the subjects after treatment with 25 mg aprocitentan, placebo, and placebo/moxifloxacin, respectively).
Mild to moderate headache was reported most frequently, followed by nasal congestion, nausea, and peripheral edema. Peripheral edema was solely reported by subjects (20.9%) after treatment with 100 mg aprocitentan. All TEAEs resolved by the end of the safety follow-up period.
Following multiple doses of 25 and 100 mg aprocitentan, an apparent dose-related decrease in hemoglobin, hematocrit, red blood cells, and platelets was noted that returned to baseline levels at the end of each period. Small changes in AST, ALT, uric acid, and cholesterol were seen in particular for the 100 mg dose that was not considered clinically relevant. Other hematology and clinical chemistry variables did not indicate study treatment-related effects.
Decreases in systolic and diastolic BP and an increase in pulse rate were observed that were greater after treatment with aprocitentan when compared to placebo or placebo/moxifloxacin. These changes were not dose-dependent or clinically relevant. Body weight appeared to increase dose-dependently after treatment with aprocitentan but no TEAE was recorded for any aprocitentan dose.
A dose-related increase in QTcF was observed after treatment with aprocitentan. Other ECG variables did not show relevant differences among study treatments. Most ECG abnormalities reported were incidental, most of them already present at baseline, and not clinically relevant. One subject after treatment with 100 mg aprocitentan presented with a QTcF change from baseline of 67 ms which was reported as an AE and resulted in discontinuation. Additional (re)tests showed that values returned within the normal range.
Discussion
This TQT study investigated the effect of aprocitentan on cardiac repolarization at concentrations associated with a dose of 25 mg (the highest approved therapeutic dose) and 100 mg aprocitentan (supratherapeutic dose). The study indicated that multiple doses of 25 mg aprocitentan o.d. (steady-state Cmax of 3.68 µg/mL) did not impact QTcF and that a prolongation above 10 ms could be excluded in the concentration range associated with the dose. At the supratherapeutic dose of 100 mg o.d., (steady-state Cmax of 16.77 µg/mL) the threshold of regulatory concern was exceeded. C-QT modeling indicated that the predicted 2-sided 90% CI exceeded the 10 ms threshold at a concentration of 16.10 µg/mL aprocitentan.
The effect of the positive control moxifloxacin was in line with published data and the by-time-point analyses confirmed the increase in QTcF following 100 mg o.d. aprocitentan.
Aprocitentan was generally well tolerated and PK was in line with those observed in previous studies. 20 With the adoption of ICH E14 in 2005, assessment of QT prolongation by systemic new clinical entities has proven a valuable tool for regulatory authorities to assess their proarrhythmic potential.40,41 The ICH E14 guidance defines a threshold of regulatory concern: a drug-induced effect on QTc beyond the upper bound limit of the 90% CI of 10 ms. The effect on QTc is evaluated as the mean placebo-corrected change in ΔΔQTc measured over a dosing interval. This provides the framework for studies in which the therapeutic and supratherapeutic doses, placebo, and a positive control drug, typically moxifloxacin, are tested in a crossover or parallel-group design.40,41 Such studies typically require large numbers of subjects and are therefore a substantial drug development investment. 42 As usually the therapeutic dose is not yet established during the early stages of drug development, traditional TQT studies are often conducted in parallel to phase 2 or 3 studies. As part of the revision of the E14 Q&A document (R3) in 2015, c-QTc analysis has emerged as an additional method to investigate drug-related effects on QTc. 35 In short, in contrast to the “by-time-point” analysis, data from all doses and time points are used to pair plasma concentrations to their effects on QTc.34,37,43,44 This considerably increases the statistical power of the c-QTc analysis, thus this approach can be implemented in studies in which the number of subjects is relatively small, such as FIH studies.34,37,43,44
In the FIH study of aprocitentan, 24-h continuous ECGs were recorded at steady-state conditions to investigate the proarrhythmic potential employing a by-time-point as well as c-QTc analysis. 25 This study did not unequivocally provide clarity on the potential of aprocitentan to impact QTc, mainly due to a low number of subjects and increased variability, therefore a waiver to avoid a full TQT study could not be obtained. Hence, the TQT study, which was performed in parallel to phase 3, was a single-center, randomized, double-blind (for aprocitentan), multiple-dose, 4-way crossover study in healthy male and female subjects. While most of the chosen design characteristics are typical (eg, choice of healthy subjects, choice of comparator, crossover design) a multiple-dose investigation was chosen. Aprocitentan has an AI of approximately 3 at steady-state conditions and its PK was not dose-proportional after single-dose administration. 25 In order to achieve multiples of therapeutic plasma concentrations a dose of 600 mg aprocitentan would need to be given as a single dose. As the intended marketed formulation of aprocitentan has a dose strength of 25 mg, it was deemed undesirable for subjects to receive that many dose units (ie, 24) in one administration.
The results of the TQT study indicated that aprocitentan at the highest therapeutic dose of 25 mg o.d. (Cmax = 3.68 µg/mL) did not prolong QT and that there was no clinically relevant effect on QTcF. However, at multiple oral doses of the supratherapeutic dose of 100 mg o.d. (Cmax = 16.77 µg/mL), the 10 ms threshold of regulatory concern was exceeded, which was both seen in the c-QT and in the by-time-point analysis. Based on c-QT modeling, the 10 mg threshold was predicted to be exceeded at an aprocitentan concentration of 16.10 µg/mL. There was no imbalance in the incidence of categorical outliers between aprocitentan and placebo.
To further evaluate the implications of the TQT study findings on aprocitentan's effect on cardiac repolarization, it is important to understand the range of concentrations that could be observed in a clinical setting in a so-called “worst case scenario.” Recently, Brussee et al published a population PK model of aprocitentan based on data from more than 900 healthy subjects and patients that were collected during phases 1 to 3. 45 The model indicated that exposure was the most impacted in subjects with low estimated glomerular filtration rate (eGFR) (AUCτ increased by 23% and 35% and Cmax increased by 19% and 30% for 30 and 17 mL/min/1.73 m2, respectively) and moderate hepatic impairment (22% higher AUCτ and 30% higher Cmax). 45 Other covariates in the model such as body weight, food status, or sex did not substantially impact PK. 45 Simulation of a high clinical exposure scenario, that is, a subject with low body weight, moderate hepatic impairment, low eGFR, and taking aprocitentan in the fed state, indicated that exposure at steady-state was higher with a relative AUCτ of 2.18 and Cmax of 2.05; that is, a 25 mg dosing regimen in the extreme scenario would yield exposures equivalent to 50 mg aprocitentan administered to a reference subject. 45
In the current described TQT study, the measured Cmax of the 25 mg dose at steady state is 3.68 µg/mL. At the recommended therapeutic dose of 12.5 mg aprocitentan used in patients, the Cmax associated with this dose is estimated to be 1.84 µg/mL, based on the fact that aprocitentan displays dose-proportional PK in the dose range up to 100 mg o.d. 20 Assuming the aforementioned relative factor, the Cmax in a high-exposure scenario is expected to be around 7.3 and 3.6 µg/mL, for 25 and 12.5 mg aprocitentan, respectively, giving margins of at least 2- and 4-fold when compared to the regulatory threshold of 16.10 µg/mL. Based on these considerations, the risk of QT prolongation with therapeutic doses of aprocitentan is considered low.
Conclusions
At the approved therapeutic doses of 12.5 and 25 mg, aprocitentan does not prolong QT to a clinically relevant extent. In contrast, supratherapeutic doses of 100 mg aprocitentan were shown to exceed the threshold of regulatory concern of 10 ms. However, as in high-exposure scenarios the Cmax is expected to be around 50% to 75% below the concentration at which the threshold of regulatory concern would be exceeded, the risk of QT prolongation with aprocitentan at therapeutic dose levels is considered low.
Footnotes
Acknowledgments
The study was conducted at CRS Clinical Research Services Mannheim GmbH, Mannheim, Germany, and was funded by Idorsia Pharmaceuticals Ltd. The principal investigator of the study was Armin Schultz and the coinvestigator was Jolanta Wierdak. ECGs were extracted by the ECG core laboratory Nabios GmbH, Munich, Germany, under supervision of Valentin Demmel. The authors thank Andreas Krause, Anne-Sophie Guern, Denis Boutin, Egle Rugyte, Giancarlo Sabbatini, Magda Fontes, Martin Wojcik, Susanne Globig, and Racheal Rowles for their contributions to the study. Patricia N. Sidharta, Janneke M. Brussee, and Jasper Dingemanse were employees of Idorsia Pharmaceuticals Ltd at the time of study conduct and analysis.
Author Contributions
PNS and JD designed, evaluated, and reported the study. JMB evaluated and reported the study. AS was the principal investigator and JW was the coinvestigator. Both clinically reviewed the data and were involved in reporting the study.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.