
Abstract
Diabetes is a long-term chronic disease, and cardiovascular disease is the leading cause of death. Diabetic cardiomyopathy (DCM), one of the cardiovascular complications of diabetes, has many uncertain factors. Epicardial fat, as the heart fat bank, functions as fatty tissue and is the heart’s endocrine organ. The existence of diabetes affects the distribution of heart fat and promotes the secretion of adipokine. In different pathological conditions, it can promote the secretion of pro-inflammatory adipokine, reactive oxygen species, oxidative stress, and even autophagy, thus affecting cardiac function. In this paper, we will elaborate on the mechanism of epicardial fat in the pathogenesis of diabetic cardiomyopathy.
Introduction
Diabetic cardiomyopathy (DCM) has been discovered for nearly 60 years. However, due to the complexity of pathogenesis, there is no specific treatment for DCM. According to current studies, diabetic cardiomyopathy is a myocardial-specific microvascular complication independent of coronary artery disease, valvular disease, hypertension, and other heart diseases. It is one of the leading causes of death in patients with diabetes. 1 DCM has a long subclinical stage, and diastolic cardiac dysfunction occurs in the early stage. At this stage, patients mostly have no clinical manifestations. However, the heart structure has changed, and the development of the late stage of the disease will involve cardiac systolic function, leading to heart failure. Initially, it was found that epicardial adipose tissue (EAT) was associated with heart failure, with preserved ejection fraction (HFpEF) having specific mutual influences. 2 At present, EAT, as an endocrine organ of the heart, has been proven to be associated with obesity, 3,4 metabolic syndrome, 5,6 type 2 diabetes, 7 atrial fibrillation, 8 coronary atherosclerotic heart disease, 9 and other diseases. In addition, some studies have shown that EAT may be used to evaluate insulin resistance and sensitivity. At present, there are few studies on epicardial adipose tissue in DCM. This article mainly elaborates on the relationship between DCM and EAT and the pathological mechanism.
Basic Knowledge of Epicardial Adipose Tissue
Epicardial adipose tissue (EAT), a visceral adipose tissue (VAT) component, originated from visceral pleural mesoderm and consisted of adipocytes, immune cells, ganglion, and inflammatory cells. Epicardial adipose tissue contains more precursor adipose cells, so its volume is smaller than other visceral and subcutaneous adipose tissues. 10 In general, more giant fat cells secrete more anti-inflammatory factors and fewer proinflammatory factors under normal physiological conditions. In addition, EAT contains more mesenchymal stem cells and is more sensitive to the generation of adipose tissue than other adipose tissues. Mesenchymal stem cells (MSCS) are the embryo’s primary source of precursor cardiomyocytes. They can develop into cardiomyocytes but lose this potential as they differentiate into adipocytes with age.
Under normal physiological conditions, EAT has a protective effect on the heart. (1) It buffers the heart from external pressure injury and mechanically protects coronary vessels; (2) One of the primary sources of cardiac energy supply; (3) Secretion of anti-inflammatory adipokines; (4) Buffering fatty acid toxicity, and so on (Figure 1). Compared with other VAT, EAT has higher absorption and release free fatty acid (FFA) ability. The oxidative metabolism of FFA absorbed from the outside world by EAT provides energy for the myocardium. The oxidation of FFA can provide 50% to 70% energy supply for the myocardium, and EAT is the main source of FFA supply for the myocardium. 11 Therefore, EAT is one of the energy supply sources for the heart and has a role in buffering lipotoxicity. There are different opinions on the mechanism of FFA to myocardial transport in EAT. The possible mechanism is the diffusion of free fatty acids to the myocardium via a concentration gradient. Alternatively, EAT secretes vasoactive substances to regulate coronary vascular tension and transfer FFA to the myocardium. 12 Nevertheless, in general, the exact mechanism needs to be clarified.

Epicardial adipose tissue’s basic knowledge.
EAT is located between the myocardium and the visceral pericardium, directly adjacent to the myocardium without fascia. Moreover, blood is supplied by coronary vessels. As the endocrine organ of the heart, EAT is the main source of adipokine. Through paracrine or vascular secretion, it can secrete leptin, adiponectin, tumor necrosis factor-alpha (TNF-α), interleukins (IL), and other cytokines that directly act on coronary vessels and myocardium. 13 EAT has a dual effect on the heart. EAT has a protective effect on the heart under physiological conditions, described in detail above. In some pathological conditions, EAT will increase in volume and thickness, resulting in increased secretion of pro-inflammatory factors and decreased secretion of cardiac protective factors such as anti-inflammatory factors, leading to the dominance of harmful effects of EAT. It is known that EAT is significantly correlated with coronary atherosclerotic heart disease, atrial fibrillation, metabolic syndrome, diabetes, and so on. In this review, we systematically expounded the relationship between DCM and EAT, hoping to promote the development of DCM further.
Epicardial Adipose Tissue and Diabetes Mellitus
Due to its characteristics, EAT has a lower glucose utilization rate than other VAT and subcutaneous adipose tissue and does not require high glucose. Diabetes Mellitus (DM) results in elevated plasma glucose levels due to insufficient insulin secretion or insulin resistance. The thickness and volume of EAT in DM patients were significantly thicker than in non-DM patients under the same conditions. 14,15 Moreover, a recent meta-analysis further confirmed the relationship between type 2 diabetes mellitus and EAT, clearly pointing out that the thickness of EAT in diabetic patients is thicker than that in nondiabetic patients. 7 Several studies have shown that taking GLP-1 agonists, 14,16,17 SGLT2-i, 16,18 metformin, 19 and other hypoglycemic drugs and insulin under the skin 20 can reduce the thickness of EAT in patients with type 2 diabetes, which further indicates that EAT is closely related to type 2 diabetes.
Epicardial adipose tissue interacts with insulin resistance. Insulin resistance (IR), as one of the main pathological mechanisms of type 2 diabetes mellitus (T2DM), is commonly used as a simple evaluation of the insulin resistance index (HOMA-IR). In the study of Gunes et al,21 homA-IR was used as an evaluation method of insulin resistance. It was concluded that EAT was positively correlated with insulin resistance and could be used as an independent predictor of insulin resistance. It was also pointed out that the optimal critical value of EAT for predicting insulin resistance was >3.85 mm. 21 However, the mechanism of EAT thickening and insulin resistance is still unclear. On the one hand, insulin resistance leads to increased EAT. Insulin resistance patients always maintain a high blood insulin level, which can promote the synthesis of fatty acids and triglycerides and increase the synthesis of adipose tissue and protein, which may be one of the reasons for the increase in EAT. On the other hand, the increase in EAT affects the occurrence of insulin resistance. Decreased GLUT4 in fat cells can lead to impaired production of branched fatty acids esters of hydroxy fatty acids (FAHFAs). FAHFAs can enhance insulin stimulation of glucose transport and insulin secretion. 15 On the other hand, as part of VAT, EAT can increase fatty acid intake, secrete inflammatory factors such as TNF, IL-6, and IL-8, antagonize insulin molecules and reduce adiponectin levels in a pathological state. In this way, islet function is impaired, and insulin sensitivity is reduced to accelerate the occurrence of insulin resistance. 22
Type 1 diabetes mellitus (T1DM), an autoimmune disease, is characterized by a progressive loss of pancreatic beta cell function and even increased blood sugar due to insufficient insulin secretion. Due to the absolute lack of insulin, the synthesis and breakdown of adipose tissue in type 1 diabetes mellitus are reduced and increased, resulting in emaciation and weight loss in these patients. However, multiple studies 23,24 have found that the epicardial fat thickness of type 1 diabetes patients is higher than that of non-diabetic patients and is independent of BMI, age, gender, and other factors. For one thing, insulin resistance may be involved in type 1 diabetes. In addition, complications associated with dyslipidemia, such as high-density lipoprotein 25 or metabolic syndrome, 26 are one of the reasons for the increase of EAT in type 1 diabetes. Nevertheless, the exact mechanism is unclear. The association between increased EAT and the risk of cardiovascular disease in type 1 diabetes patients is unclear. Svanteson et al 27 found that patients with type 1 diabetes had an increased risk of coronary heart disease, independent of EAT. Zobel et al, 28 as far as the relationship between epicardial fat diabetic patients with myocardial microvascular study, found that diabetes epicardial fat thickness is higher than that of a control group, but in people with type 1 diabetes, it is far lower than in patients with type 2 diabetes. The change of epicardial adipose tissue and the relationship between diabetic myocardial microvascular do not evident. In conclusion, the increase in EAT of type 1 diabetes patients is due mainly to the combination of high-risk factors such as metabolic syndrome and insulin resistance. In terms of current research progress, the influence of the disease on the EAT increase is not known, and the relationship between the disease and cardiovascular disease is unclear.
Plasma retinol-binding protein 4 (RBP4) is an adipocytokine secreted and expressed by adipose tissue. 29 The decrease of GLUT4 in adipocytes leads to the increase of serum retinol-binding protein RBP4 secreted by adipocytes, and the increase of serum RBP4 level leads to impaired glucose uptake by skeletal muscle and increased glucose production by the liver. In contrast, increased serum RBP4 level significantly reduces insulin sensitivity. 30 Meanwhile, extensive epidemiological studies have demonstrated that RBP4 is a circulating biomarker of insulin resistance and the risk of cardiovascular disease and metabolic syndrome. 31 -33 In 2021, Chauhan et al further proved that RBP4 could be used as an early evaluation indicator of type 2 diabetes and was linearly correlated with HOMA-IR. With increased serum RBP4 level of 1 mg/L, HOMA-IR increased significantly by 0.007 units. 34 In 2021, Chauhan et al further proved that RBP4 could be used as an early evaluation indicator of type 2 diabetes and was linearly correlated with HOMA-IR. We also need to know that the increase of RBP4 is a partly toll-like receptor 4 (TLR4) mediated proinflammatory effect. NLRP3 inflammasome is induced to release interleukin-1β (IL1β) glucose-dependent through TLR4/MD2 receptor complex to promote adipose tissue inflammation and impair insulin sensitivity. 35,36 In addition, higher levels of RBP4 and lower levels of GLUT4 were found in EAT, which can increase the harm of cardiovascular diseases. 37 Studies have shown that 38 serum RBP4 positively correlates with the Gensini score, which can reflect the severity of coronary arteries to a certain extent. More importantly, RBP4 and NT-proBNP have the same value in predicting the prognosis of chronic heart failure. 39 It is further suggested that there are numerous links between EAT, insulin resistance, and cardiovascular diseases, which is worth further exploration.
Epicardial Adipose Tissue and Diabetic Cardiomyopathy
DCM refers to cardiac diseases in patients with diabetes and is independent of hypertension, coronary heart disease, valvular heart disease, and other heart diseases. 40 The basis of DCM is the existence of diabetes, and the interaction between diabetes and EAT is known above. There are few studies on DCM and EAT. However, DCM is closely related to EAT from the pathogenesis of DCM, including metabolic abnormalities (glucotoxicity and lipid toxicity), inflammation and oxidative stress, abnormal activation of neurohumoral fluid, and dysfunction of coronary artery and vascular circulation.
Epicardial Adipose Tissue and Abnormal Glucose and Lipid Metabolism
Diabetes often leads to glycolysis and gluconeogenesis abnormalities, impairing heart function by reducing glucose uptake by cells and producing energy from glucose metabolism. As a kind of heterotopic fat pool, the fat accumulation of EAT is an important factor in obesity-related insulin resistance and inflammation. Adipose tissue absorbs very little glucose, accounting for only 10% of the dietary sugar load. Compared with other VAT and subcutaneous adipose tissue, EAT has a very low utilization rate of glucose, which is related to the need to maintain a high lipid metabolism level of EAT. As the only glucose-lowering hormone in the body, insulin has a very low distribution in EAT and lower sensitivity, which is related to the fact that the presence of insulin can inhibit fat metabolism. Smith and Kahn proposed that glucose transporter 4 (GLUT4), which is responsible for glucose transport and glucose uptake in fat cells, can be used as one of the indicators to evaluate insulin sensitivity. 15 Moreover, GLUT4 expression and glucose metabolism in adipocytes modulate insulin sensitivity by altering endocrine function, substrate metabolism, new fat production, or release of novel FAHFA lipids. Therefore, what role GLUT4 of seemingly weak fat cells plays in EAT is worthy of further study. Early studies have shown that the mRNA expression level of GLUT4 in EAT is lower than that in subcutaneous adipose tissue, thus aggravating cardiac insulin resistance. 37 It is assumed that abnormal GLUT4 of EAT leads to increased cardiac insulin resistance. The myocardium suffers from high glucose injury, aggravating mitochondrial dysfunction, and inflammatory factor release and oxidative stress aggravate cardiac dysfunction. In the study of Naryzhnaya et al, 41 the production degree of insulin-dependent reactive oxygen species (ROS) and the inhibition degree of insulin-dependent lipid hydrolysis were used as indicators to evaluate insulin sensitivity. It was found that decreased insulin sensitivity and increased ROS production was positively correlated with EAT adipocyte hypertrophy, which resulted in vascular endothelial dysfunction, aggravated inflammation, and eventually serious aggravation of coronary heart disease and other cardiovascular diseases. Greulich et al 42 further demonstrated the causal relationship between EAT and insulin resistance. They found that epicardial fat secreted SMAD2 phosphorylation (A marker of activin A signaling) in mice fed a high-fat diet, which reduced the insulin-mediated phosphorylation of Akt-Ser473, thereby inducing insulin resistance. In addition, 2 prospective studies have found that dapagliflozin 43 or metformin 44 can reduce epicardial fat thickness, improve the differentiation of EAT cells, and improve the occurrence of insulin resistance, further proves that hypertrophy of EAT adipocytes and increase EAT thickness will lead to aggravated insulin resistance, myocardial mitochondrial dysfunction, increased oxidative stress, and increased release of inflammatory factors, and ultimately impair cardiac function. Of course, cardiac insulin resistance and systemic insulin resistance are related or can be said to interact with each other, co-regulation.
Due to the decrease in glucose energy metabolism, the intake of fatty acids in diabetic patients will increase to replenish energy for the body through fatty acid β oxidation and tricarboxylic acid cycle metabolism of fatty acids. This change is compatible with the physiological requirement of EAT. However, cardiomyocytes and non-adipose cells have limited capacity to store fat. When they exceed the body’s compensation level, fatty acid accumulation will occur in cells, resulting in cellular steatosis 45 and even cellular dysfunction and apoptosis, which will also affect mitochondrial function and inhibit autophagy, leading to cardiac dysfunction. 46 Epicardial adipocytes are characterized by reduced expression of genes that regulate lipid metabolism, particularly lipoprotein lipase, which depends on the size of the adipocyte, which leads to fat decomposition in hypertrophic adipocytes. 47
Epicardial Adipose Tissue and Inflammatory Reaction
As an endocrine organ of the heart, EAT can secrete anti-inflammatory adipokines to protect the heart under physiological conditions. However, under the stimulation of pathological conditions, it can manifest in the volume of adipocytes or thickness of EAT and the expression of pro-inflammatory adipocytokines such as TNF, IL-1, and IL-6 aggravates the inflammatory response of the heart. The increased expression of TNF-α can induce the aggregation and infiltration of macrophages and stimulate the release of inflammatory factors, thus leading to the oxidative stress of mitochondria and endoplasmic reticulum, the production of ROS or reactive nitrogen species (RNS) and accelerating the process of apoptosis, and ultimately impaired heart function. 11 At the same time, studies have shown that EAT can produce more ROS products, express more intense oxidative stress, and lower catalase levels than other adipose tissues, such as subcutaneous or visceral tissue. 48
Macrophages are the main effector cells of the inflammatory response, which can phagocytose, secrete and kill microorganisms to maintain body homeostasis. Scavenger receptors (SRs) determine the type of macrophage differentiation. EAT inflammatory reaction shows a high concentration of infiltrating white blood cells, mainly T lymphocytes, macrophages, and inflammatory cytokines, 49 and EAT inflammatory reaction can be expressed through macrophage infiltration. 50 In a normal body, M1 macrophages recruit various inflammatory cells to fight pathogens and play the function of immune surveillance. M2 macrophages promote angiogenesis and tissue repair and play a role in inhibiting inflammation. In the pathological state, M1 macrophages are overly active, the inflammatory reaction is aggravated, and many inflammatory factors such as IL-1b, IL-6, and TNF-α are secreted. In contrast, the overactivation of M2 macrophages is mainly manifested as excessive tissue repair, leading to fibrosis and other changes. The activation of macrophages may be related to the expression of scavenger receptors in adipocytes. Martin-Fuentes et al 51 found that different types of scavenger receptors, such as CD36, macrophage scavenger receptor 1 (MSR1), lectin-like oxidized LDL receptor 1 (LOX-1), and placental collector 1 (CL-P1), affect the activation of different types of macrophages. This finding was again demonstrated in 2019 by Santiago-Fernandez et al, 52 who found increased expression of LOX-1, CL-P1, CD68, and CD11c scavor receptors in patients with ischemic myocardium and diabetes compared with patients with non-ischemic cardiomyopathy or non-diabetes, increased infiltration of M1 macrophages.
Increased scavenger receptor expression aggravates the progression of cardiovascular disease. It has been reported that increased expression of scavenger receptors can lead to increased expression of inflammatory cytokines, 53 thereby aggravating the progression of the disease. Hirata et al 54 EAT macrophages were significantly infiltrated, the proportion of M1/M2 macrophages increased, the expression of proinflammatory factors increased, and the coronary artery lesions became severe. This phenomenon is significant in patients with diabetes. 55 Macrophages are believed to express scavenger receptors through adipocytes to mediate inflammatory responses essential in cardiovascular disease. In addition, the cell signaling pathway may be another mechanism of macrophage-related inflammation. Huang et al 56 found that inflammatory reactions can activate and infiltrate macrophages by activating the NF-κB pathway of EAT. Gurses et al 57 showed that Netrin-1 could also mediate the polarization of EAT macrophages. As a vascular endothelial factor, Netrin-1 can play an inflammatory role by activating the PI3K-AKT signaling pathway, Wnt signaling pathway, and other pathways. In addition, activation of M1 macrophages may be related to the AMPK pathway 58 or Toll-like receptor 59 activation. However, which signaling pathway is needed to mediate it requires more research.
Adiponectin, an endogenous anti-inflammatory adipocytokine, is an insulin sensitizer that can improve insulin resistance and inhibit the production and release of TNF. Both high glucose levels and inflammatory response can reduce adiponectin expression in EAT, thus enhancing the release of TNF, IL-6, and other inflammatory factors by EAT endothelial cells. 9,60 Antonopoulos et al 61 demonstrated that adiponectin expression in EAT is controlled by the paracrine action of oxidative products released by the heart. Moreover, adiponectin can reduce the activity of nicotinamide adenine dinucleotide phosphate oxidase (NOX)-2. Nox-2, as an important enzyme mediator in the myocardial inflammatory response and ROS and RNS production, can enhance myocardial inflammatory response on the one hand; on the other hand, it can lead to increased production of TNF-β and other inflammatory factors, which also suggests that adiponectin may play a role in inhibiting TNF production through NOX-2. All of the above will lead to myocardial fibrosis and cardiac diastolic dysfunction, which are the pathological changes characteristic of DCM in the early stage. In addition, reduced adiponectin leads to impaired mitochondrial oxidative phosphorylation in EAT, 62 leading to mitochondrial and cardiac dysfunction. Li et al 19 demonstrated in 2020 that metformin can increase adiponectin expression signal and decrease TNF-α, TNF-β1, and IL-6 expression, thereby improving atrial fibrosis and atrial remodeling. Adiponectin may play an important role in DCM and may be a breakthrough point for future treatment.
Epicardial Adipose Tissue and Abnormal Neurohumoral Regulation
There have been few studies on the correlation between EAT and RAAS in recent years. In 2008, a study on the relationship between the RAAS of EAT and insulin resistance drew increasing attention. In this study, patients undergoing cardiac surgery were found to have some degree of insulin resistance after surgery. Angiotensinogen mRNA expression in epicardial adipose tissue was significantly increased from baseline. In contrast, angiotensinogen mRNA expression in subcutaneous adipose tissue remained unchanged, and surgery did not affect the associated receptors. It suggests that the increase in angiotensinogen may be related to insulin resistance. 63
The RAAS axis in the body consists of 2 pairs of vertical axes. One is the well-known axis of the main physiological function—angiotensin-converting enzyme, Angiotensin ⅱ type 1-receptor (AT1-R) axis; Angiotensin ⅱ type 2-receptor (AT2-R) axis. This axis exerts its effects mainly through the combination of AT1 R: vasoconstriction, water, sodium retention, sympathetic nerve stimulation, and so on. The other axis is the less-mentioned ACE-2-mediated hydrolysis of Ang ⅱ, which ultimately produces ANG1-7 binding to THE Mas receptor to produce an anti-AT1-R effect. 64 What we need to mention this time is the ACE2-ANG1-7 axis. In 2016, Patel et al 65 found that ACE2-deficient mice had more inflammation of EAT than wild-type mice. They detected decreased myocardial adiponectin, increased cardiac steatosis and lipotoxicity, and increased myocardial insulin resistance, thus worsening cardiac function. These changes could be reversed by infusion of Ang 1-7 into ACE2 mice. In the same year, Professor Vaibhav B Patel 66 made a detailed summary of the role of the ACE2-ANG1-7 axis in EAT and found that ACE2 was expressed in adipose cells and up-regulated in mice induced by the high-fat diet. Moreover, ACE2 deficiency leads to diastolic cardiac insufficiency partly due to EAT inflammatory response. In addition, the decomposition product of ACE2, ANG1-7, improves cardiac dysfunction mainly through adiponectin expression and EAT inflammatory attenuation. Therefore, ACE2-Ang1-7 is considered to be a key regulator of EAT.
In 2017, Prof. Marcel Blumensatt led the team to find that Ang ⅱ expression level increased 2.6 times in T2DM-EAT medium and cardiomyocytes cultured together compared with those cultured without T2DM or EAT. At the same time, the expression of renin and AT1 R increased, and ACE2 decreased through RAS paracrine action. Such changes lead to decreased mitochondrial β oxidation and shortened myocardial sarcomere. 67 These results suggest that RAAS activation of EAT may induce the occurrence of diabetic cardiomyopathy. However, there are few studies on RAAS between DCM and EAT, and more studies are needed to prove it in the future. Besides, we need to know that the leptin-aldosterone-neprilysin Axis is mentioned. Leptin receptors can activate the sympathetic nervous system and renin-angiotensin system and stimulate aldosterone secretion. The interaction between leptin and aldosterone enhances the renal sympathetic nerve, resulting in adrenalin activity. This complex neurohormonal interaction leads to poor ventricular remodeling and cardiac fibrosis. Changes in the number and biology of epicardial adipose tissue further promote the release of leptin and other pro-inflammatory adipokines, leading to cardiac and systemic inflammation and terminal organ fibrosis. 68 Perhaps this unique neuromodulation axis can become a therapeutic target and breakthrough point in the future.
Epicardial Adipose Tissue and Autophagy
Autophagy is a highly conserved catabolic process that degrades unnecessary or dysfunctional intracellular wastes through the action of lysosomes and plays an important role in stabilizing cell homeostasis. 69 Autophagy plays an important role in cardiac development, protecting the adaptive changes of cardiac myocytes, coping with external stress, and contributing to cardiac plasticity. Autophagy plays a dual role in DCM. On the one hand, in type 2 diabetes, hyperglycemia, hyperlipidemia, oxidative stress, and other stimuli inhibit autophagy by inhibiting the AMPK pathway and enhancing the mTOR signaling pathway of cardiomyocytes. On the other hand, an impaired insulin signaling pathway in T2DM leads to autophagy overactivation in the diabetic heart. 70 Autophagy dysfunction induces mitochondrial oxidative stress, abnormal protein accumulation, and aggravated inflammatory response to damage cardiac function. Although the activation of autophagy contributes to the recovery of cardiac myocyte function, it is still necessary to know that the over-activation of autophagy in DCM can lead to the self-digestion of cells and enhance the production of ROS. In addition, preclinical tests have proved that autophagy in DCM mice is dysfunctional. 71 Therefore, both autophagy overactivation and/or autophagy inhibition of DCM can lead to cardiac changes related to myocardial fibrosis, myocardial cell apoptosis, and cardiac dysfunction.
Autophagy is a key regulator of adipogenesis in brown adipose tissue. 72 EAT is believed to have similar functions and structures to brown fat, with smaller fat cells, a high concentration of mitochondria, and the compensatory function of protecting the body. Sacks et al 73 found that Uncoupling Protein (UCP)-1, as a specific marker and medium of brown fat cell thermogenesis, was significantly more than 5 times more expressed in EAT than retrosternal fat. At the same time, UCP-1 was almost undetected in subcutaneous fat, indicating that EAT may be a brown adipose tissue. However, brown adipose tissue gradually disappears with age. It is rarely distributed in the adult body, mainly in the neck and back, armpit, mediastinum, and other parts that produce more heat. Autophagy is involved in transforming white fat to brown fat, and the autophagy function is enhanced with the increase of brown fat. According to our conjecture, the thickening of EAT, as brown fat, is related to the disorder of autophagy. Cairo and Villarroya 74 helped us verify that dysregulation of adipocyte autophagy leads to adipose tissue thickening, but the adipose tissue in this study was not brown adipose tissue. As a negative regulator of autophagy, Rubicon is knocked out in adipocytes, resulting in atrophy and lipid accumulation in adipocytes, 75 which seems to contradict the previous research results. However, the loss of autophagy will accelerate apoptosis, 76 and autophagy has 2 sides in DCM, so more studies are needed to confirm how autophagy plays its role in the EAT of DCM. In addition, we still need to know that metformin treatment may improve cardiac function by down-regulating NLRP3 inflammasome and autophagy, leading to decreased levels of fat tissue pro-inflammatory markers, such as TNFα, IL6, and mitochondrial ROS production. 77 In a word, autophagy may be a potential therapeutic target for future research.
Epicardial Adipose Tissue and Exosome
Exosomes are vesicle-like bodies secreted by cells with a lipid bilayer membrane structure, which contain various organelles and bioactive substances such as RNA, lipids, and proteins. They are widely found in plasma, saliva, myocardium, inflammation, and other tissues. Exosomes communicate with ligands through intercellular integration molecules, adhesion molecules, and four-molecule cross-linking families. The miRNA delivered by exosomes can target mRNA expression in recipient cells, thereby regulating recipient cells’ phenotype and protein expression. 78 Epicardial fat contains abundant mesenchymal stem cells, which are cardiac precursors during fetal development and become adipocytes in adulthood. As a member of the adipose-derived stem cell secretome, exosomes can drive the heterogeneous expression of stem cells, including inflammation and cell proliferation, which provides the possibility for cardiomyocyte regeneration (Figure 2). 79,80
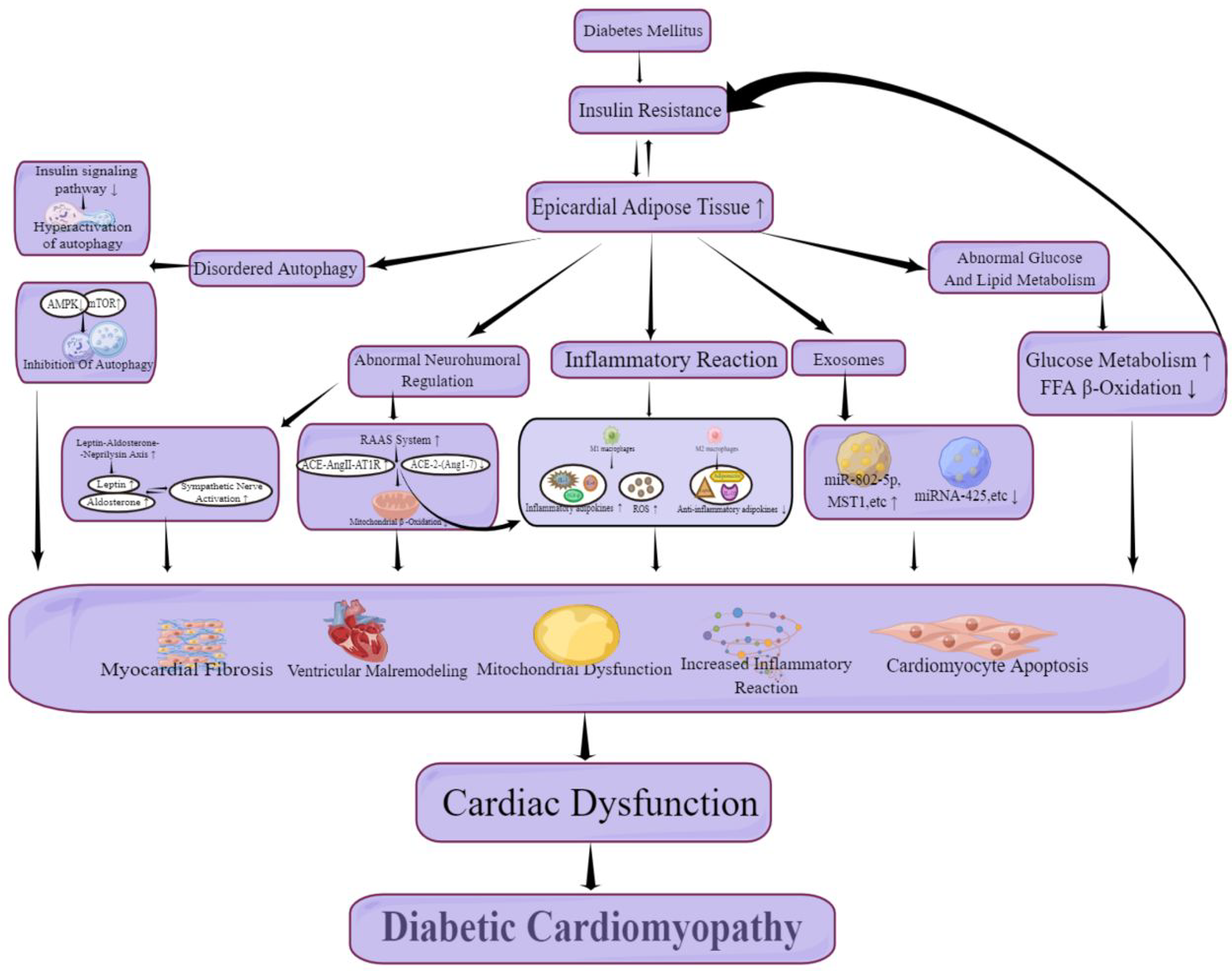
Pathogenesis of diabetic cardiomyopathy. The relationship of diabetes mellitus, epicardial adipose tissue and cardiac dysfunction. AMPK indicates adenosine monophosphate activated protein kinase; mTOR, mammalian target of rapamycin; RAAS, rennin-angiotensin-aldosterone system; ACE, angiotensin converting enzyme; Ang, angiotensin II type 1 receptor; ROS, reactive oxygen species; IL, interleukin; TNF, tumor necrosis factors; FFA, free fatty acid.
Exosomes may be involved in the pathogenesis of DCM, but the specific mechanism remains unclear. It has been reported that exosomal microRNA can regulate pancreatic β-cell activity by regulating the intercellular signaling cascade, which is an essential determinant of pancreatic β-cell function integrity. 81,82 Exosomes derived from hypertrophic adipocytes miR-802-5p can cause insulin resistance decreased adiponectin, and enhanced oxidative stress by down-regulating HSP60. 83 By cultivating mice with diabetic cardiomyopathy, Wang et al 84 found that the epicardial adipose tissue of such mice released some harmful exosomes, resulting in cardiac hypertrophy, apoptosis, and cardiac dysfunction. Hu et al 85 found that MST1, as a harmful exosome, can promote the development of diabetic cardiomyopathy by inhibiting GLUT4 translocation, inhibiting glucose uptake in patients with diabetes mellitus, increasing insulin resistance, inhibiting cardiomyocyte autophagy, and promoting cardiomyocyte apoptosis. Notably, in 2021, Shaihov-Teper et al 86 found that exosomes from epicardial fat had a higher inflammatory response and myocardial fibrosis in patients with atrial fibrillation. However, exosomal miRNA-425 can inhibit the progression of myocardial fibrosis and cardiac dysfunction by binding to the 3 ‘untranslated region of TGF-β1. 87 In conclusion, exosomes are involved in developing diabetes, insulin resistance, inflammation aggravation, autophagy function inhibition, myocardial fibrosis, and other cardiac dysfunction. Exosomes also have a myocardial protective effect. Therefore, the impact of diabetic cardiomyopathy may be 2-fold, which needs more studies to clarify (Figure 2).
Conclusions
Diabetic cardiomyopathy is a complex problem in clinical treatment because of its long course and non-specific clinical manifestations. We found that the complexity of the pathogenesis of diabetic cardiomyopathy is closely related to epicardial adipose tissue, whether it is abnormal glucose and lipid metabolism and activation of the neurohumoral system, or severe inflammatory reaction, inhibition of autophagy function and diversity of exosomes. Epicardial adipose tissue, as the “protective layer” and endocrine organ of the heart, maybe a potential therapeutic target of DCM.
Footnotes
Author Contributions
Conceptualization, method, writing-manuscript, visualization: Xueyuan Yang; writing-review and editing: Chao Feng; guidance: Jinping Feng.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.