
Abstract
A high platelet-to-lymphocyte ratio (PLR) has recently been associated with ischemic outcomes in cardiovascular disease. Increased platelet reactivity and leukocyte-platelet aggregate formation are directly involved in the progress of atherosclerosis and have been linked to ischemic events following percutaneous coronary intervention (PCI). In order to understand the relation of PLR with platelet reactivity, we assessed PLR as well as agonist-inducible platelet aggregation and neutrophil-platelet aggregate (NPA) formation in 182 acute coronary syndrome (ACS) patients on dual antiplatelet therapy with aspirin and prasugrel (n = 96) or ticagrelor (n = 86) 3 days after PCI. PLR was calculated from the blood count. Platelet aggregation was measured by multiple electrode aggregometry and NPA formation was determined by flow cytometry, both in response to ADP and SFLLRN. A PLR ≥91 was considered as high PLR based on previous data showing an association of this threshold with adverse ischemic outcomes. In the overall cohort and in prasugrel-treated patients, high PLR was associated with higher SFLLRN-inducible platelet aggregation (67 AU [50-85 AU] vs 59.5 AU [44.3-71.3 AU], P = .01, and 73 AU [50-85 AU] vs 61.5 AU [46-69 AU], P = .02, respectively). Further, prasugrel-treated patients with high PLR exhibited higher ADP- (15% [11%-23%] vs 10.9% [7.6%-15.9%], P = .007) and SFLLRN-inducible NPA formation (64.3% [55.4%-73.8%] vs 53.8% [44.1%-70.1%], P = .01) as compared to patients with low PLR. These differences were not seen in ticagrelor-treated patients. In conclusion, high PLR is associated with increased on-treatment platelet reactivity in prasugrel-treated patients, but not in patients on ticagrelor.
Keywords
Introduction
Platelets and leukocytes play key roles in the perpetuation and enhancement of inflammation, in particular in atherosclerosis. They provide numerous cytokines and growth factors for their interplay. 1,2 For instance, this mutual influence is apparent as high counts of platelets adhering to leukocytes are seen in acute myocardial infarction, but also in many chronic inflammatory diseases. 3 Neutrophils are the most abundant type of white blood cells in the circulation and a crucial part of the response to acute inflammation. In atherosclerotic processes, they infiltrate plaques and play a major role in plaque destabilization and rupture. 4
Platelet-to-lymphocyte ratio (PLR) is an easily-obtainable marker comprising the patient's platelet count and specifics of the white blood cell count, that is, the number of lymphocytes. An elevated PLR has been associated with in-hospital and long-term adverse events in patients with acute coronary syndromes (ACS). 5 -7 Moreover, high PLR has been associated with poor overall survival in patients with acute heart failure 8 and predicted target vessel restenosis in patients undergoing infrainguinal angioplasty with stent implantation or percutaneous coronary intervention (PCI). 9,10
High on-treatment residual platelet reactivity (HRPR) in response to ADP and SFLLRN has been associated with adverse ischemic events following angioplasty and stenting. 11 In addition, leukocyte-platelet aggregates are increased in patients after myocardial infarction. 12
In order to understand the relation of PLR with on-treatment platelet reactivity and leukocyte-platelet aggregate formation, we assessed platelet aggregation and neutrophil-platelet aggregate (NPA) formation in 182 patients undergoing PCI with stent implantation for ACS.
Methods
Study Population
The study population consisted of 182 ACS patients on daily aspirin (100 mg/day), and either prasugrel (10 mg/d, n = 96), or ticagrelor (180 mg/d, n = 86) therapy (Figure 1). Blood sampling was performed 3 days after successful acute PCI after an overnight fast. Exclusion criteria were a known aspirin, prasugrel or ticagrelor intolerance (allergic reactions, gastrointestinal bleeding), a therapy with vitamin K antagonists (warfarin, phenprocoumon, acenocoumarol) or direct oral anticoagulants (rivaroxaban, apixaban, dabigatran, edoxaban), treatment with ticlopidine, dipyridamol or nonsteroidal anti-inflammatory drugs, a family or personal history of bleeding disorders, malignant myeloproliferative disorders or heparin-induced thrombocytopenia, severe hepatic failure, known qualitative defects in platelet function, a major surgical procedure within 1 week before enrollment, a platelet count <100.000 or >450.000/µL and a hematocrit <30%.
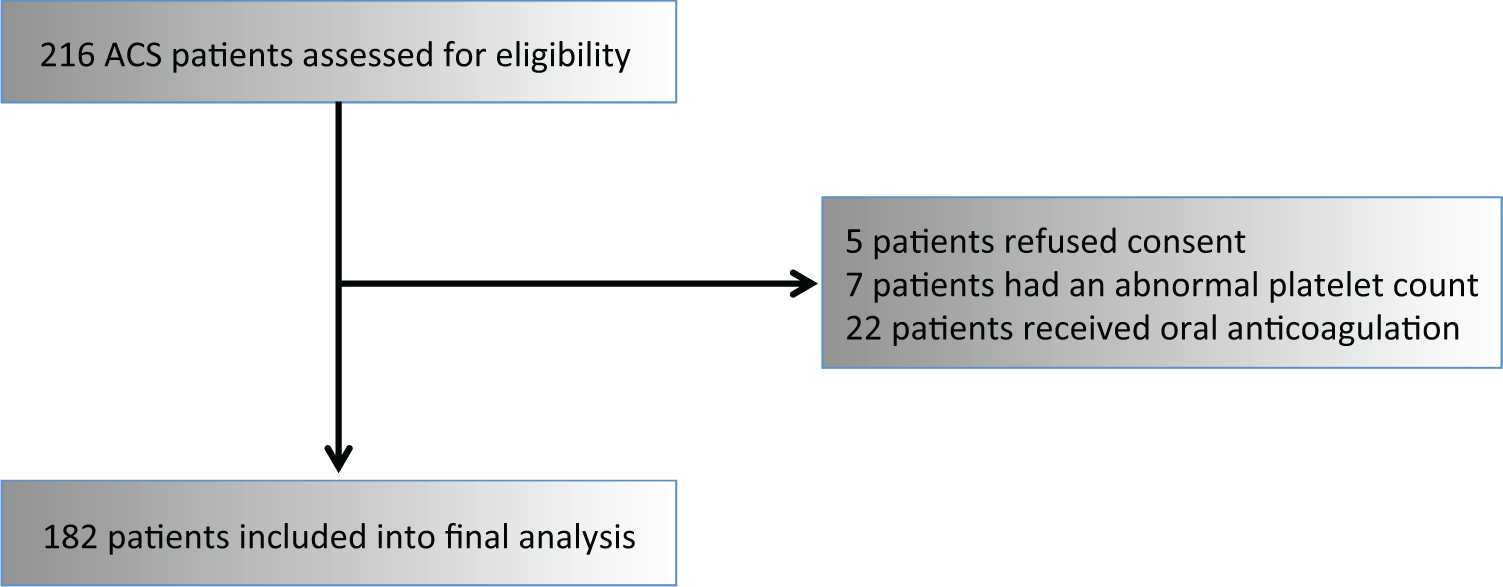
Flow chart illustrating patient enrollment.
The study protocol was in accordance with the Declaration of Helsinki and its later amendments and approved by the Ethics Committee of the Medical University of Vienna. All study participants gave their written informed consent.
Measurement of Platelet, Neutrophil, and Lymphocyte Count
The hemogram was measured on the Sysmex Xe-5000 System, Sysmex Corporation, Japan, in the central laboratory of the Medical University of Vienna according to standardized protocols.
Multiple Electrode Aggregometry
Whole blood impedance aggregometry was performed with the Multiplate analyzer (Roche Diagnostics, Mannheim, Germany) as previously described. 13,14 One Multiplate test cell contains 2 independent sensor units and 1 unit consists of 2 silver-coated highly conductive copper wires with a length of 3.2 mm. After dilution (1:2 with 0.9% NaCl solution) of hirudin-anticoagulated whole blood and stirring in the test cuvettes for 3 minutes at 37°C, adenosine diphosphate (ADP, 6.4 μM, Roche Diagnostics, Mannheim, Germany) or SFLLRN (protease activated receptor [PAR]-1 agonist, 32μM, Roche Diagnostics, Mannheim, Germany), was added and aggregation was continuously recorded for 6 minutes. The concentrations of all agonists were chosen according to the manufacturer's recommendations. The adhesion of activated platelets to the electrodes led to an increase of impedance, which was detected for each sensor unit separately and transformed to aggregation units (AU) that were plotted against time. The AU at 6 minutes were used for calculations. One AU corresponds to 10 AU*min (area under the curve of AU).
NPA Formation
NPA were measured as previously published with small modifications. 15,16
In brief, HEPES buffer, or ADP (1.5 μM), or SFLLRN (7.1 µM) were added to 5 µL whole blood, which had been diluted with 55 µL HEPES-buffered saline. The concentrations of all agonists were determined in previous titration experiments with increasing dosages of each agonist in 10 healthy controls. The selected concentrations of agonists induced about 60% to 70% of the maximal achievable increase in NPA formation in healthy controls. After 10 minutes, monoclonal antibodies (anti-CD45-peridinin chlorophyll protein (Becton Dickinson (BD)), anti-CD41-phycoerythrin, (Immunotech, Marseilles, France), and anti-CD14-allophycocyanin, BD), or isotype-matched controls were added. After 15 min, samples were diluted with FACSlysing solution and at least 10.000 CD45+ events were acquired immediately. Neutrophils were identified based on their side scatter versus CD14 characteristics, and NPAs were determined by recording CD45+CD41+ events (Figures S1-S3).
Statistical Analysis
Statistical analysis was performed using the Statistical Package for Social Sciences (SPSS version 24.0, SPSS, Chicago, Illinois, USA). Median and interquartile range of continuous variables are shown. Categorical variables are given as number (%). We performed the non-parametric Mann Whitney U tests to detect differences in continuous variables. The chi-square test was used to assess differences in categorical variables. Spearman rank correlation was used to assess correlations.
Multivariable linear regression analyses using a backward elimination algorithm with a P value ≤.1 for removal were used to adjust for patient characteristics. Adjustment was performed for the following variables after thoughtful variable selection: age, sex, 17,18 body mass index, 13,19 type of P2Y12 inhibitor, type of ACS, arterial hypertension, diabetes mellitus, hyperlipoproteinemia, smoking, history of myocardial infarction, creatinine, and high-sensitivity C-reactive protein (hsCRP).
Two-sided P values <.05 were considered statistically significant. HRPR was defined according to a recent consensus document. 20 The respective cut-off value for HRPR ADP by MEA were AU >46. 20
The cut-off value for PAR-1 mediated platelet aggregation was derived from a group of 55 healthy Caucasian volunteers (male/female 21/34; aged 42 ± 13 years), who served as control population in a previously published study. 21 The corresponding cut-off value were AU ≥71 for normal PAR-1 mediated platelet aggregation (SFLLRN as agonist) by MEA. 21,22 The cut-off value for high PLR was a PLR ≥91, according to a previous study linking this threshold with target vessel restenosis (TVR) following angioplasty and stenting for peripheral artery disease. 9
Results
Clinical, laboratory and procedural characteristics of the study population are given in Table 1. Patients on ticagrelor treatment were older and had higher creatinine levels than those treated with prasugrel. No other significant differences were observed.
Clinical and Laboratory Characteristics of Prasugrel- and Ticagrelor-Treated Patients.a

Abbreviations: BMI, body mass index; ACE, angiotensin converting enzyme; CRP, C-reactive protein; MI, myocardial infarction; TIA, transient ischemic attack; WBC, white blood cell count.
a Continuous data are shown as median (interquartile range). Dichotomous data are shown as n (%).
Agonist-inducible platelet aggregation and NPA formation is depicted in Table 2. Platelet aggregation in response to ADP and SFLLRN did not differ between patients on prasugrel and patients on ticagrelor. Of note, HRPR based on the ADP consensus cut-off value of AU >46 20 was seen in only 2 patients, both on prasugrel.
Platelet Aggregation by MEA and NPA Formation in Response to ADP and SFLLRN in Prasugrel- and Ticagrelor-Treated Patients.

Abbreviations: ADP, adenosine diphosphate; AU, aggregation units; MEA, multiple electrode aggregometry; NPA, neutrophil-platelet aggregates.
Based on the previously published cut-off value of AU ≥71, 21 high SFLLRN-inducible platelet aggregation was present in 72 patients (40%). Of these patients, 41 and 31 received prasugrel and ticagrelor, respectively (42.7% of prasugrel-treated patients vs 36% of ticagrelor-treated patients; P = .36).
NPA formation in response to ADP was similar in prasugrel- and in ticagrelor-treated patients. NPA formation in response to SFLLRN was significantly higher in patients on prasugrel than in those on ticagrelor (Table 2). Unstimulated levels of NPA are given in Table 2.
In the overall study population, ADP-inducible platelet aggregation correlated with ADP-inducible NPA formation (r = 0.16, P = .03). This correlation was due to the correlation in prasugrel-treated patients (r = 0.37, P < .001), while no significant correlation was seen in ticagrelor-treated patients (r = −.02, P = .83).
Likewise, there was a correlation between SFLLRN-inducible platelet aggregation and SFLLRN-inducible NPA formation in the overall study population (r = 0.23, P = .002), which was again due to the correlation in prasugrel-, but not in ticagrelor-treated patients (prasugrel: r = 0.25, P = .01; ticagrelor: r = 0.19, P = .08).
In the overall study population ADP- and SFLLRN-inducible platelet aggregation correlated with each other (r = 0.58, P < .001), which was similar in prasugrel- (r = 0.57, P < .001) and ticagrelor-treated patients (r = 0.61, P < .001). Likewise, ADP- and SFLLRN-inducible NPA formation correlated in the overall study population (r = 0.44, P < .001) as well as in prasugrel- and ticagrelor-treated patients (both r = 0.42, both P < .001).
A high PLR (PLR ≥ 91 9 ) was seen in 136 (75%) patients in the overall study population. PLR differed significantly between prasugrel- and ticagrelor-treated patients (104.6 [86.6-137.3] vs 120 [96.7-155.3], P = .006; Figure 2). After dichotomization, high PLR was seen in 66 (69%) of prasugrel-treated patients and in 70 patients (81%) on ticagrelor (P = .05).

Platelet-to-lymphocyte ratio (PLR) in prasugrel- vs ticagrelor-treated patients.
Patients with high PLR were older than those with low PLR (58.6 years [51.6-67 years] vs 54.6 years [47.6-62.2 years], P = .048). There was no association of creatinine levels with high or low PLR in the overall study population (data not shown). There were no significant differences in age or serum creatinine between patients on prasugrel (P = .05 and P = .43) and ticagrelor (P = .71 and P = .52) with high or low PLR (data not shown).
In univariate analyses, PLR did not correlate with ADP- or SFLLRN-inducible platelet aggregation, but with ADP-inducible NPA formation, due to the significant correlation in prasugrel-treated patients (Table 3). PLR did not correlate with SFLLRN-inducible NPA formation, even if patients on prasugrel and ticagrelor were evaluated separately (Table 3).
Correlations of PLR With MEA and NPA Formation in Response to ADP and SFLLRN in the Overall Study Population as well as in Prasugrel- and Ticagrelor-Treated Patients.

Abbreviations: ADP, adenosine diphosphate; AU, aggregation units; MEA, multiple electrode aggregometry; NPA, neutrophil-platelet aggregates; PLR, platelet-to-lymphocyte ratio.
In multivariable linear regression analyses adjusting for confounders PLR was associated with ADP-inducible NPA formation (β = 0.214, P = .009) and with SFLLRN-inducible platelet aggregation (β = 0.208, P = .013), whereas no association of PLR with ADP-inducible platelet aggregation and SFLLRN-inducible NPA formation was detectable (Table 4).
Association of PLR With MEA and NPA Formation in Response to ADP and SFLLRN in the Overall Study Population Adjusting for Confounders.

Abbreviations: ADP, adenosine diphosphate; AU, aggregation units; MEA, multiple electrode aggregometry; NPA, neutrophil-platelet aggregate; PLR, platelet-to-lymphocyte ratio.
Next, we were interested to evaluate if either high PLR or low PLR values were linked to on-treatment platelet aggregation or NPA formation.
Patients with high and low PLR had similar levels of ADP-inducible platelet aggregation (20 AU [15-24 AU] vs 18.5 AU [14.8-23 AU]; P = .27). Also, ADP-inducible platelet aggregation was similar in prasugrel-treated patients with high vs low PLR (20 AU [15-23 AU] vs 18.5 AU [15-22.3 AU]; P = .9) and in those on ticagrelor with high vs low PLR (20 AU [15-25 AU] vs 17.5 AU [12.5-23.8 AU]; P = .23, respectively).
In contrast, high PLR was linked to higher SFLLRN-inducible platelet aggregation compared to low PLR (67 AU [50-85 AU] vs 59.5 AU [44.3-71.3 AU], P = .01). This observation in the entire cohort was attributable to the results obtained in prasugrel-treated patients, who had higher SFLLRN-inducible platelet aggregation in case of high PLR as compared to low PLR (73 AU [50-85 AU] vs 61.5 AU [46-69 AU]; P = .02; Figure 3). No such difference was discernible in patients on ticagrelor (high PLR: 65 AU [48.8-82.3 AU] vs low PLR 53 AU [37.5-74.8 AU]; P = .12; Figure 3).

Platelet aggregation by multiple electrode aggregometry (MEA) and neutrophil-platelet aggregate (NPA) formation in response to SFLLRN in prasugrel- (A and B) and ticagrelor- (C and D) treated patients with high versus low platelet-to-lymphocyte ratio (PLR).
NPA formation in response to ADP did not differ significantly between patients with high and low PLR in the overall study population (13.3% [10.3%-17.9%] vs 10.9% [7.9%-18.7%], P = .06). However, in prasugrel-treated patients, we observed a significant difference between those with high vs those with low PLR regarding the formation of NPA in response to ADP (15% [11%-23%] vs 10.9% [7.6%-15.9%], P = .007). No such difference was seen in patients on ticagrelor (high PLR 12.8% [9.4%-16.3%] vs low PLR 12.9% [9.2%-19.1%], P = 7).
High and low PLR were not significantly associated with NPA formation in response to SFLLRN in the overall study population (high PLR 62.6% [52.3%-70.7%] vs low PLR 53.6% [42.6%-69.7%], P = .06). Still, in patients on prasugrel, high PLR was associated with higher SFLLRN-inducible NPA formation (64.3% [55.4%-73.8%] vs 53.8% [44.1%-70.1%]; P = .01; Figure 3). No such association was detected in ticagrelor-treated patients (61.4% [45.1%-68%] vs 53.6% [35.6%-66.2%]; P = .39; Figure 3).
Discussion
To the best of our knowledge, this is the first study associating on-treatment platelet aggregation and NPA formation with PLR in ACS patients treated with prasugrel or ticagrelor. We demonstrate that PLR was independently associated with ADP-inducible NPA formation and SFLLRN-inducible platelet aggregation. Moreover, patients with high PLR had higher SFLLRN-inducible platelet aggregation than patients with low PLR. Finally, prasugrel-treated patients with high PLR exhibited higher ADP- and SFLLRN-inducible NPA formation as compared to patients with low PLR.
High PLR has repeatedly been identified as predictor of adverse outcomes in patients receiving dual antiplatelet therapy after cardiac and peripheral stenting. 9,23 Moreover, ST-elevation myocardial infarction (STEMI) patients with high PLR exhibited larger infarct sizes and a higher rate of no-reflow phenomenon compared to patients with low PLR. 13 Also, PLR has been associated with the extent of coronary and carotid artery disease, 24,25 and repeatedly linked to adverse outcomes in ACS patients. 26,27 Further, high PLR was associated with disease severity and longer hospitalizations during the current pandemia in patients infected with SARS-CoV-2. 28,29
Potent platelet inhibition is a mainstay in the pharmacological treatment of patients with atherosclerotic disease, 30 particularly when undergoing stent implantation, and on-treatment residual platelet reactivity has been numerously linked to adverse outcomes. MEA is an easily-obtainable and highly standardized platelet function test, which has been related to outcome data in clopidogrel-treated patients. 20 On the other hand, inflammatory processes are pivotal for the development of atherosclerosis and pro-inflammatory states have been associated with negative outcomes in patients with overt CAD. 31 We measured NPA formation because neutrophil-platelet interaction and neutrophil density have previously been associated with impaired coronary microcirculation, myocardial damage and left ventricular dysfunction in patients with STEMI. 32 Neutrophils can augment coagulation and atherosclerosis by inducing thrombin generation via neutrophil extracellular traps (NETs), leading to platelet activation and the development of ischemic events. 33 -35 The important interplay between platelets and leukocytes in the development of atherosclerosis is possibly reflected in PLR and may therefore make it a new valuable prognostic marker in atherosclerosis. 36,37 Indeed, we found a significant association of PLR with ADP-inducible NPA in our cohort.
Current antiplatelet therapy with the novel P2Y12 inhibitors has been shown to be superior to clopidogrel in ACS patients by exerting a more potent inhibition of ADP-inducible platelet aggregation. 38,39 This fact might explain the lack of association of PLR with ADP-inducible platelet aggregation in our cohort, contrary to a previous report demonstrating a significant association of PLR with high residual platelet reactivity (defined as change in maximal aggregation ≤20% from baseline aggregation in response to ADP) in patients treated with clopidogrel. 40 However, platelet activation via other surface receptors remains largely unaffected. Thrombin acts as a strong endogenous platelet agonist activating platelets predominantly via PAR-1. 41 -44 PAR-1 activation can occur despite potent P2Y12 inhibition, 21,22,45 and we have previously shown that PAR-1 mediated platelet activation predicts long-term ischemic events in patients with lower extremity artery disease. 11,21,22 This is of particular interest, as PLR was associated with SFLLRN-inducible platelet aggregation in our cohort and, hence, high PLR might reflect a prothrombotic milieu and explain the association of PLR with adverse outcomes in previous reports. 9,23 Also, we have identified a number of patients with high SFLLRN-inducible platelet activation despite adequate P2Y12 inhibition, which may explain the stronger associations of PLR with PAR-1 mediated platelet activation as compared to ADP-inducible platelet activation. Of note, the addition of the PAR-1 inhibitor vorapaxar to antiplatelet therapy decreased ischemic events in patients with CAD, but significantly increased bleeding risk and has therefore never been included in the standard antiplatelet regimen. 46,47
The differences in the associations of platelet aggregation and NPA formation with PLR observed between prasugrel- and ticagrelor-treated patients may be due to different pharmacokinetics and –dynamics of the thienopyridine prasugrel and the cyclopentyl-triazolopyrimidine ticagrelor. 48 Ticagrelor is proposed to bind to a second pocket consisting of transmembrane segments and the extracellular loop 2 as well as the N-terminal domain of P2Y12. 49 This binding site is suggested as a mechanism for platelet inhibition independent of ADP. 49 Further biological effects have been attributed to ticagrelor, which are different from prasugrel and possibly not due to P2Y12 inhibition, like the inhibition of cellular adenosine uptake and of toll-like receptor 1/2 mediated platelet activation. 50 -52 The missing association of PLR with NPA formation in ticagrelor-treated patients suggests that ticagrelor may at least in part overrule platelet activation via pathways that are independent of P2Y12 inhibition. 52 There is a dispute, however, whether ticagrelor or prasugrel are more beneficial 50,53 -55 with a non-significantly more favorable 1-year outcome in ticagrelor-treated patients as described by Motovska et al. 55 and a significantly more favorable 1-year outcome for patients treated with prasugrel in the Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment (ISAR-REACT) 5 study. 56
Limitations
A study limitation is the lack of clinical outcome data. Further, a potential selection bias exists due to the choice of the P2Y12 antagonist by the treating physician.
Whole blood methods to determine platelet function may differ from results obtained by assays that use separated platelets. However, whole blood assays are considered advantageous to assays using separated platelets, as the process of separation may affect platelet response to agonists. Moreover, whole blood assays better reflect the in vivo conditions of each individual, as the interplay of all blood components is preserved. For the reasons mentioned above, we did not use light transmission aggregometry (LTA), even though it can be considered the historical gold standard of platelet function testing. In initial studies we have seen that results are affected at platelet counts <100.000 and >450.000/µL and a hematocrit <30%. We therefore did not include individuals with these abnormal laboratory values. In detail, 7 patients were not enrolled due to an abnormal platelet count at screening (Figure 1).
We cannot completely exclude potential laser coincidence events related to platelet counts. However, by flow cytometry, we assessed each sample first without agonists and then after agonist-induced activation for comparison, staining specifically with a platelet marker.
Conclusion
In conclusion, our study demonstrates the association of on-treatment platelet aggregation and NPA formation with PLR in prasugrel-treated ACS patients. We therefore propose further evaluation of PLR as simple, readily-available and inexpensive marker of a prothrombotic state.
Supplemental Material
Supplemental Material, sj-pdf-1-cpt-10.1177_10742484221096524 - Platelet-to-Lymphocyte Ratio as Marker of Platelet Activation in Patients on Potent P2Y12 Inhibitors
Supplemental Material, sj-pdf-1-cpt-10.1177_10742484221096524 for Platelet-to-Lymphocyte Ratio as Marker of Platelet Activation in Patients on Potent P2Y12 Inhibitors by Patricia P. Wadowski, Joseph Pultar, Constantin Weikert, Beate Eichelberger, Maximilian Tscharre, Renate Koppensteiner, Simon Panzer and Thomas Gremmel in Journal of Cardiovascular Pharmacology and Therapeutics
Supplemental Material
Supplemental Material, sj-pdf-2-cpt-10.1177_10742484221096524 - Platelet-to-Lymphocyte Ratio as Marker of Platelet Activation in Patients on Potent P2Y12 Inhibitors
Supplemental Material, sj-pdf-2-cpt-10.1177_10742484221096524 for Platelet-to-Lymphocyte Ratio as Marker of Platelet Activation in Patients on Potent P2Y12 Inhibitors by Patricia P. Wadowski, Joseph Pultar, Constantin Weikert, Beate Eichelberger, Maximilian Tscharre, Renate Koppensteiner, Simon Panzer and Thomas Gremmel in Journal of Cardiovascular Pharmacology and Therapeutics
Supplemental Material
Supplemental Material, sj-pdf-3-cpt-10.1177_10742484221096524 - Platelet-to-Lymphocyte Ratio as Marker of Platelet Activation in Patients on Potent P2Y12 Inhibitors
Supplemental Material, sj-pdf-3-cpt-10.1177_10742484221096524 for Platelet-to-Lymphocyte Ratio as Marker of Platelet Activation in Patients on Potent P2Y12 Inhibitors by Patricia P. Wadowski, Joseph Pultar, Constantin Weikert, Beate Eichelberger, Maximilian Tscharre, Renate Koppensteiner, Simon Panzer and Thomas Gremmel in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Author Contributions
Patricia P. Wadowski: Patient recruitment, data analysis, and writing the initial draft; Joseph Pultar: Patient recruitment, critical revision, and final approval; Constantin Weikert: Patient recruitment, critical revision, and final approval; Beate Eichelberger: Laboratory measurements, critical revision, and final approval; Maximilian Tscharre: Data analysis, critical revision, and final approval; Renate Koppensteiner: Critical revision and final approval; Simon Panzer: Study design, writing the initial draft, critical revision, and final approval; Thomas Gremmel: Study design, data analysis, writing the initial draft, critical revision, and final approval.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research was funded by the “Medical Scientific Fund of the Mayor of the City of Vienna,” grant number 14016, and by the “Anniversary Fund of the Austrian National Bank,” grant number 16155, to Thomas Gremmel.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.