
Abstract
Background:
Duchenne muscular dystrophy (DMD) is a neuromuscular disease caused by dystrophin gene mutations affecting striated muscle. Due to advances in skeletal muscle treatment, cardiomyopathy has emerged as a leading cause of death. Previously, nicorandil, a drug with antioxidant and nitrate-like properties, ameliorated cardiac damage and improved cardiac function in young, injured mdx mice. Nicorandil mitigated damage by stimulating antioxidant activity and limiting pro-oxidant expression. Here, we examined whether nicorandil was similarly cardioprotective in aged mdx mice.
Methods and Results:
Nicorandil (6 mg/kg) was given over 15 months. Echocardiography of mdx mice showed some functional defects at 12 months compared to wild-type (WT) mice, but not at 15 months. Disease manifestation was evident in mdx mice via treadmill assays and survival, but not open field and grip strength assays. Cardiac levels of SOD2 and NOX4 were decreased in mdx vs. WT. Nicorandil increased survival in mdx but did not alter cardiac function, fibrosis, diaphragm function or muscle fatigue.
Conclusions:
In contrast to our prior work in young, injured mdx mice, nicorandil did not exert cardioprotective effects in 15 month aged mdx mice. Discordant findings may be explained by the lack of cardiac disease manifestation in aged mdx mice compared to WT, whereas significant cardiac dysfunction was previously seen with the sub-acute injury in young mice. Therefore, we are not able to conclude any cardioprotective effects with long-term nicorandil treatment in aging mdx mice.
Introduction
Cardiomyopathy is a leading cause of death in patients with Duchenne muscular dystrophy (DMD), a progressive muscle wasting disease. 1 -4 Despite advances in therapies for DMD, specific treatments for DMD cardiac failure have been limited. 5 -7 Studies have begun to unravel the cellular processes contributing to pathology in DMD cardiomyopathy. These studies have investigated potential treatments such as nitric oxide (NO) donors, histone deacetylase (HDAC) inhibitors, and anti-fibrotic agents. 6 -9 While these therapies have shown the capacity to improve cardiac function in mdx mice, this has not translated to human studies. To effectively develop treatments for DMD cardiomyopathy, it is important to have model systems that can recapitulate human DMD cardiac disease. This has been a major limitation for the field. While natural history studies have examined the disease manifestations in DMD animal models, 10 -12 therapeutic interventions in these models remain understudied. Traditional heart failure medications such as angiotensin-converting enzyme (ACE) inhibitors, beta-blockers and corticosteroids have been used for the management of DMD cardiomyopathy, but these drugs are unable to halt the progression of the disease or repair pre-existing damage. 13 Therefore, there is a clear need to develop precise and effective treatments for DMD cardiomyopathy.
One potential drug that has recently demonstrated beneficial effects in the DMD heart is nicorandil, vasodilatory drug most often used to treat angina. 14 -16 Nicorandil is a pleotropic drug that modulates NO signaling, 17 acts as an antioxidant, 14,18,19 and improves mitochondrial health by stimulating kATP channel activity. 20 Since deficiencies in these cellular processes have been ascribed to DMD cardiac pathology, 21 -24 nicorandil represents an attractive therapeutic option warranting further study.
Previously, nicorandil has exhibited cardioprotective effects in in vitro and in vivo models of DMD cardiomyopathy. 14,15 Mechanistically, nicorandil treatment was shown to be cardioprotective in vitro by reducing oxidative stress and protecting mitochondria, resulting in decreased cardiomyocyte injury and cell death. In vitro, nicorandil exerted these effects by stimulating kATP channels and antioxidant capacity. 14 In vivo, nicorandil was shown to protect mdx hearts from fibrotic injury through enhancing antioxidant activity as evidenced by increased SOD2 levels, and diminished nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) and xanthine oxidase (XO) activity. 15
Here, we investigated whether nicorandil would be cardioprotective long-term against the cardiomyopathy that can naturally occur in aging mdx mice. 25 Young, 11-week old female mdx mice were aged to 15 months to allow for the DMD-associated cardiac defect to manifest and nicorandil supplementation began at 3 months. Mild mdx cardiac dysfunction was observed by echocardiography at 12 months, but was absent at 15 months. No change in cardiac function was seen with nicorandil treatment. Similarly, no histological change in cardiac fibrosis was observed with nicorandil. Echocardiographic analysis of diaphragm function demonstrated mdx impairment at 12 months and continuing at 15 months and this was unchanged by nicorandil. In open field and grip strength testing, mdx mice performed comparably to WT at 9, 12 and 15 months and performance was not altered with nicorandil treatment. Treadmill testing indicated muscle fatigue at 9, 12 and 15 months that was not improved with nicorandil. Lastly, protein levels of SOD2 and NOX4 were diminished in mdx hearts compared to WT, and these were unchanged by nicorandil treatment. Since aging mdx mice did not develop a significant cardiac defect by 15 months, we are unable to conclude any beneficial effect of long-term nicorandil treatment on the heart.
Results
Long-Term Nicorandil Treatment Does Not Improve Cardiac or Diaphragm Function in Aging mdx Mice
To investigate whether long-term supplementation of nicorandil in mdx mice was cardioprotective, cardiac function was analyzed by echocardiography and cardiac section were analyzed by histology to assess fibrosis. Mice received fresh drinking water daily with nicorandil (6 mg/kg), beginning at age 3 months. To effectively determine the impact of nicorandil on the heart, it was important to measure cardiac function every 3 months, beginning at 9 months. Cardiac abnormalities in mdx mice have been seen as early as 1 month, with heart failure most often observed between 9 and 12 months. 10,26 WT and vehicle-treated mdx mice served as controls for comparison. Animals were aged to 15 months, at which point hearts were collected for histology and protein isolation. Western blotting confirmed dystrophin deficiency in mdx hearts and skeletal muscle (Supplementary Figure 1).
Echocardiography was performed to look for evidence of left ventricular hypertrophy to assess structural changes to the heart. 27,28 No obvious differences in cardiac function were observed between WT and mdx at 9 months (Table 1). Hearts were weighed after study conclusion at 15 months to compare to body weight as a marker of hypertrophy. Figure 1 shows an increase in the ratio of heart to body weight in mdx vs. WT, suggestive of cardiac hypertrophy. Hypertrophy does not appear to be ameliorated with nicorandil treatment. At 12 months, differences among mdx and WT respectively included: interventricular septal end diastole (IVSd; 0.087 vs. 0.071; P < .05), left ventricular posterior wall end diastole (LVPWd; 0.088 vs. 0.072; P < .05), and left ventricular mass ASE (LVD mass ASE; 0.703 vs. 0.681; P < .01), suggestive of left ventricular hypertrophy. However, these differences were no longer apparent at 15 months. Percent ejection fraction was surprisingly elevated in 12 and 15 month mdx compared to WT (12 month: 62% vs. 54%; n.s.; 15 month: 61% vs. 54%; n.s.). Nicorandil did not significantly improve the % ejection fraction in 12 month (58% vs. 62%; n.s.) or 15 month (55% vs. 61%; n.s.) mdx mice compared to vehicle. Percent fractional shortening was comparable in mdx vs. WT at 12 months (29% vs. 24%; n.s.) and 15 months (28% vs. 24%; n.s.). Nicorandil did not significantly improve fractional shortening at 12 months (26% vs. 29%; n.s.) or 15 months (25% vs. 28%; n.s.) in mdx mice. Together, there did not appear to be cardiac dysfunction in mdx hearts compared to WT, even at 15 months. Further, nicorandil did not appear to exert any cardioprotective effects against age-induced decline in heart function.
Echocardiographic Measurements of WT and mdx Mice With or Without Nicorandil Treatment.a

a The hearts of young (11 week) female mdx or WT mice were subject to echocardiography every 3 months, starting at 9 months, to evaluate functional changes with age and nicorandil treatment. Data represent mean ± SEM, two-way ANOVA, Tukey’s multiple comparisons test. Group size (N) is as noted.
b Indicates P < .05 15 months vs. 9 months.
c Indicates P < .05 mdx + vehicle vs. WT + vehicle.

Ratio of heart to final body weight in mice suggests hypertrophy. At the conclusion of the study, animals and hearts were weighed for comparison of heart to body weight ratio. Data show an increase in ratio in both mdx groups compared to WT, suggestive of cardiac hypertrophy that is unchanged with nicorandil treatment. Data represent mean ± SEM, N = 8-15/group, one-way ANOVA, ****P < .0001.
Masson trichrome staining was performed on 15 month mdx cardiac sections to evaluate whether nicorandil minimized cardiac fibrosis. Cardiac fibrosis in mdx hearts was elevated compared to WT (11.3% vs. 7.7%; P < .05; Figure 2). Long-term nicorandil treatment did not diminish mdx cardiac fibrotic area (11.3% vs. 12.6%; n.s.; Figure 2).

Nicorandil does not decrease cardiac fibrosis in 15-month aged mdx mice. Hearts from WT or mdx mice treated with vehicle or nicorandil were harvested after 15 months and 8 μM sections were cut and stained with Masson’s trichrome stain. A, Representative images show increased cardiac fibrosis present in 15-month mdx hearts vs. WT, and it is not diminished with nicorandil treatment. B, All sections stained were analyzed with Metamorph Software for % fibrotic area. Long-term treatment with nicorandil did not diminish cardiac fibrosis. Data represent mean ± SEM, N = 6-14/group, one-way ANOVA, *P < .05 mdx vs. WT, mdx vs. vehicle, n.s.
Diaphragm function was analyzed in parallel with cardiac function in order to characterize the disease status of mdx mice over time, and whether nicorandil had beneficial effects. Echocardiography was used to measure diaphragm function at 9, 12 and 15 months of age (Table 2) using a previously established protocol. 29 At 9 months, duration of inspiration (DI) is altered in mdx vs. WT (0.11 vs. 0.14; P < .05). At 12 months, mdx diaphragm impairment compared to WT is further evidenced by alterations in DI (0.12 vs. 0.17; P < .01), velocity of inspiration (VI; 0.53 vs. 0.78; P < .05) and velocity of expiration (VE; 0.53 vs. 0.81; P < .001). Impairment continues at 15 months in mdx compared to WT: diaphragm excursion distance (DED; 0.04 vs. 0.09; P < .001), DI (0.14 vs. 0.17; P < .05), VI (0.37 vs. 0.68; P < .01) and VE (0.45 vs. 0.70; P < .05). Surprisingly, animals receiving nicorandil showed a decline in DED compared to vehicle-treated mdx at 12 months (0.04 vs. 0.09; P < .0001). On all other measurements, diaphragm function between nicorandil and vehicle-treated mdx was comparable, indicating it did not improve respiratory dysfunction. Together, these data indicate that mdx mice had disease-induced decline in respiratory function and nicorandil treatment was not sufficient to improve pathological changes in diaphragmatic function.
Diaphragm Measurements in Mice.a
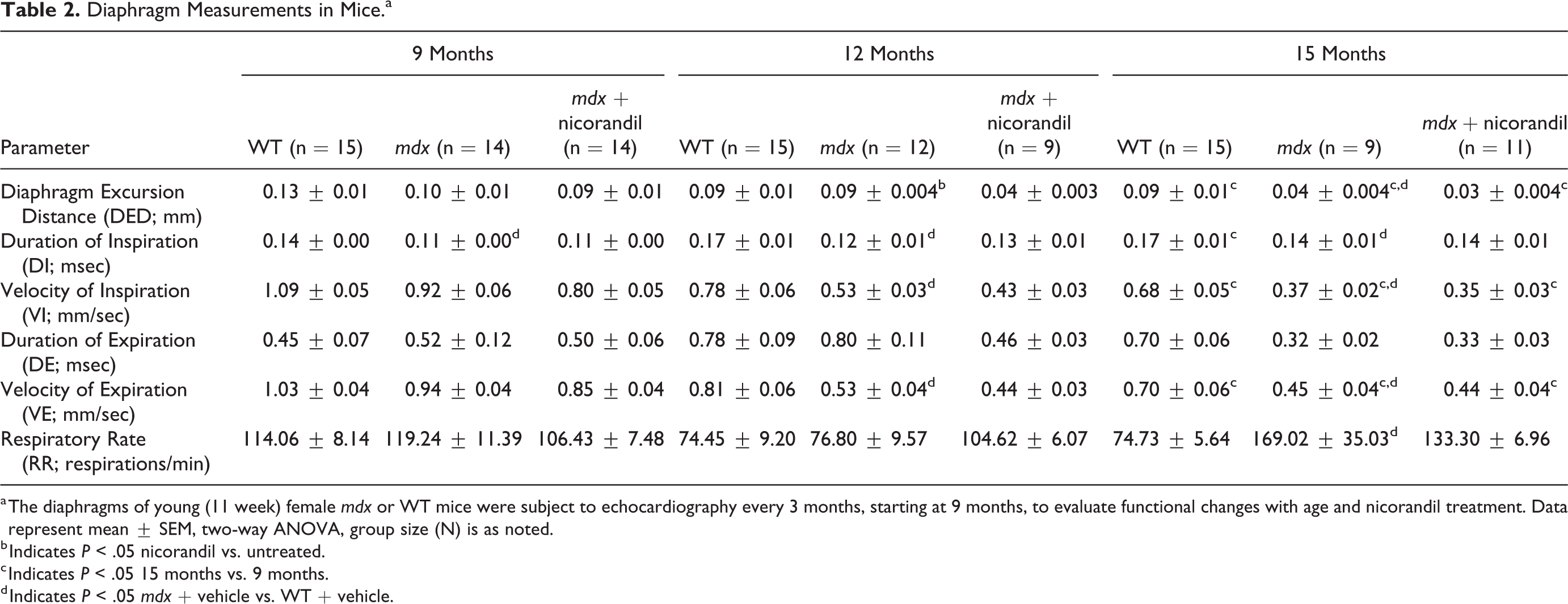
a The diaphragms of young (11 week) female mdx or WT mice were subject to echocardiography every 3 months, starting at 9 months, to evaluate functional changes with age and nicorandil treatment. Data represent mean ± SEM, two-way ANOVA, group size (N) is as noted.
b Indicates P < .05 nicorandil vs. untreated.
c Indicates P < .05 15 months vs. 9 months.
d Indicates P < .05 mdx + vehicle vs. WT + vehicle.
Nicorandil Extends the Lifespan of mdx Mice
Terminal studies were carried out until 70 weeks of age. Vehicle treated mdx mice began to succumb to premature death as early as 13 weeks (Figure 3A). At the termination of the study at 70 weeks, 100% of WT mice remained, whereas 53% of vehicle-treated mdx and 67% of nicorandil treated mice remained (n.s.). Specific group sizes at each timepoint are noted in Tables 1 and 2. Throughout the study, several mdx mice developed tumors which required euthanasia, which is a complication that has previously been reported in mdx. 30 On average, mdx mice receiving nicorandil tended to live longer compared to vehicle treated (65 weeks vs. 61 weeks; n.s.; Figure 3B). Together, these analyses indicated that mdx mice were dying prematurely of disease-related complications, and that while nicorandil treatment was associated with prolonged survival, it did not impact survival to a statistically significant extent.

Nicorandil treatment increases survival in mdx mice. A, Kaplan-Meier analysis of mouse survival throughout the 15-month experiment. All 15 WT mice survived the entirety of the study, whereas 8 mdx + vehicle mice survived, and 10 mdx + nicorandil. Nicorandil treatment did increase survival in mdx mice. B, Average lifespan of mice. Data represent mean age ± SEM, n.s., N = 8-15/group.
Nicorandil Treatment Does Not Mitigate the Effects of Skeletal Muscle Wasting in mdx Mice
We next examined whether nicorandil treatment was protective against the skeletal muscle wasting known to occur in mdx mice. 31 -33 Animals were subject to open field analysis, grip strength measurements and treadmill analysis. These behavioral analyses have been established as a robust mechanism for assessing muscle fatigue in mice. 34 -36 WT and mdx mice had no significant differences in open field testing at any time point with respect to distance traveled, mean speed, maximum speed, time spent mobile and time spent immobile (Figure 4A-E). There was a significant reduction in mdx mobile episodes in comparison to WT at 12 months (48 vs. 32; P < .05; Figure 4F). This trend in reduced mobile episodes continued at 15 months, but was not significant (47 vs. 33; n.s.). In contrast, mdx immobile episodes were also reduced at 12 months compared to WT (Figure 4G). No alteration of open field performance was observed with nicorandil treatment (Figure 4).

Nicorandil does not improve open field performance of mdx mice. Open field testing was performed to measure animal activity at 9, 12, and 15 months. Animals were subject to measuring (A) mean distance, (B) mean speed, (C) max speed, (D) time mobile, (E) time immobile, (F) mobile episodes, and (G) immobile episodes. Impaired performance of mdx vs. WT animals was observed at 12 months with respect to mobile and immobile episodes. All other data demonstrated no significant differences in performance regarding either disease status or nicorandil treatment. Data represent mean age ± SEM, two-way ANOVA, N = 8-15/group. ^Indicates P < .05 mdx + vehicle vs. WT + vehicle.
Forelimb grip strength increased with age in WT mice, whereas it remained relatively constant among vehicle or nicorandil treated mdx mice (Figure 5A). Whole limb grip strength increased with age in all treatment groups and no differences were evident among WT, mdx and nicorandil-treated (Figure 5B).

Nicorandil treatment does not alter grip strength in mdx mice. Grip strength was tested in animals to assess forelimb and whole limb muscle function over 9, 12, and 15 months and with nicorandil treatment. A, Forelimb grip strength in mdx mice was impaired at 12 and 15 months compared to WT, and this was not improved with nicorandil treatment. B, Whole limb grip strength diminished in all groups with age and was not different in mdx vs. WT or with nicorandil treatment. Data represent mean age ± SEM and was normalized to body weight, n.s., N = 8-15/group. *Indicates P < .05 15 months vs. 9 months.
Treadmill analyses demonstrated a decrease in distance traveled by mdx mice in comparison to WT at all timepoints, and nicorandil did not increase distance (Figure 6A). The duration of travel was also impacted in mdx mice, showing a significant decrease in the duration at all ages in comparison to WT (Figure 6B). No improvement in duration traveled was seen with nicorandil treatment.

Nicorandil does not improve exercise fatigue observed in mdx mice. Mice were subject to fatigue testing by treadmill at 9, 12, and 15 months of age. Distance traveled and duration of travel were assessed. A, mdx mice were fatigued sooner than WT, as indicated by a shorter distance traveled. This did not improve with nicorandil treatment. B, mdx mice also traveled for a shorter duration in comparison to WT and this was not improved with nicorandil treatment. ^Indicates P < .05 mdx + vehicle vs. WT + vehicle. #Indicates P < .05 nicorandil vs. untreated. Data represent mean ± SEM, two-way ANOVA, N = 8-15/group.
Open field mobile episodes and treadmill analyses indicated disease-induced muscle fatigue in mdx vs. WT, although no age-related decline was observed in any group. mdx mice performed comparably to WT in all other behavioral assays. It is important to note that by the final 15 month timepoint, only 8 of the original 15 mice were still remaining, and this may that of the mice that survived to advanced age (12-15 months), any DMD-disease processes that may have occurred were not evident in comparison to WT age-matched controls. Overall, long-term nicorandil administration did not appear to alter mdx performance in open field, grip strength and treadmill analyses.
Nicorandil Does Not Alter Markers of Oxidative Stress SOD2 and NOX4 in mdx Hearts
Since nicorandil has previously been shown to stimulate antioxidant activity and diminish oxidative stress in the heart, 14,15,37,38 we next examined the levels of antioxidant SOD2, as well as the levels of pro-oxidant NOX4 in 15 month mdx hearts. We sought to establish whether there were any cellular alterations that had occurred, despite the lack of functional cardiac improvement (Table 1). Protein was harvested from 15 month hearts at the conclusion of the study, and western blotting was performed to assess SOD2 and NOX4 levels. Figure 7A shows a representative blot and all data were normalized to vinculin, which served as a loading control (Figure 7B). SOD2 levels tended to be increased in WT vs. mdx hearts, indicative of an increased antioxidant capacity (Figure 7C). In animals treated with nicorandil, no changes in SOD2 levels were seen. Levels of NOX4 were diminished in mdx in comparison to WT, and unchanged with nicorandil (Figure 7D). Together, these data show that nicorandil did not appear to stimulate antioxidant activity in the heart, in a natural aging mdx model of DMD cardiomyopathy.

Long-term nicorandil treatment does not alter SOD2 and NOX4 levels in mdx hearts. Hearts were harvested from 15-month-old mice and protein was isolated for western blot analysis of SOD2 and NOX4. A, Representative blots of NOX4, SOD2, and vinculin. B, Vinculin served as a loading control for data normalization. C, SOD2 levels were slightly decreased in mdx + vehicle vs. WT, although this was not significant. Nicorandil treatment did not alter expression. D, NOX4 levels were significantly diminished in mdx + vehicle in comparison to WT. Levels did not change with nicorandil treatment. Data represent mean age ± SEM, N = 8-15/group, *P < .05 mdx + vehicle vs. mdx + nicorandil, **P < .05 WT control vs. mdx + vehicle.
Discussion
Current pharmacologic agents for the management of DMD cardiomyopathy include traditional heart failure drugs including ACE inhibitors, beta-blockers, angiotensin receptor blockers, and anti-inflammatory agents such as corticosteroids. 13 While these treatments manage the symptoms of heart failure, they are often insufficient to halt the progression of cardiomyopathy or repair existing cardiac damage. 13 As the cellular and metabolic phenotypes underlying DMD cardiomyopathy have been identified, potential treatment options for each of these have been or are currently being investigated for therapeutic potential. Studies involving exon skipping drugs and gene therapy have shown promise to improve skeletal muscle function, but few examine and report improvement in cardiac function. 39 -45 Moreover, there is supporting evidence to suggest that targeting just skeletal muscle may contribute to cardiac injury in DMD. 46 Other treatments under investigation in the DMD heart include cell-based therapies and membrane sealants, which have shown promising results and are now in clinical trials. 47 -51 Other agents being investigated for their ability to target specific cellular phenotypes have shown to stimulate nitric oxide (NO) signaling, 14,52,53 diminish oxidative stress, 54 -56 preserve mitochondrial function 57 and downregulate cell death processes. 50,51 While these molecular treatments have shown promise in animal models of DMD, they have not yet led to clinical improvement in patients with DMD cardiomyopathy.
The mdx mouse is frequently used as a model to investigate DMD cardiomyopathy. 10 However, mdx mice are known to be of limited use because they do not display the early dilated cardiomyopathy observed in human patients. 58 This model manifests the skeletal muscle disease but often does not recapitulate the severity of the human cardiac failure in DMD. 10,59 Young mdx mice may need to be aged as old as 21 months to allow cardiomyopathy to manifest. 60 Since the cardiac defect in this model is mild and often takes several months to manifest, researchers often induce stress in animals with either exercise or other pharmacologic agents such as dobutamine or isoproterenol to reveal the cardiac defect. 5,60 -63 Others have developed additional strains of the mdx mouse such as mdx4cv/mTRG2 and mdx/Cmah−/− to demonstrate a more severe cardiac phenotype. 58,64,65 Our lab has previously modeled DMD cardiomyopathy using low dose isoproterenol to induce cardiac injury and found this model sufficiently unmasks the cardiac defect present in young mdx but not WT mice. 15
Recently, we examined the effects of short-term nicorandil treatment on isoproterenol-induced cardiac injury in young mdx mice. 15 This short-term study demonstrated that nicorandil treatment in young mdx mice rescued isoproterenol-induced cardiac injury through improving % EF and FS, diminishing cardiac fibrosis and enhancing antioxidant levels (Table 3). That study served as the rationale for us to examine whether long-term nicorandil treatment could also be cardioprotective. In this study, we investigated whether long-term nicorandil treatment was cardioprotective in a natural history model of DMD cardiomyopathy, the mdx mouse. In order to demonstrate cardioprotection, we first needed to establish the time points where mdx mice began to develop cardiac dysfunction. Here, mdx mice as young as 12 months demonstrated some LV defects in comparison to WT. However, at 15 months, no differences in cardiac function were observed with respect to either age-related or disease-related decline in function (Table 3). This may be because mdx mice do not display as severe of a cardiac defect in comparison to human disease 10 and severe cardiac impairment may not be seen until >21 months of age. 25,66 Tachycardia has been previously reported in mdx mice, 67,68 but was not evident in mdx mice in this study, even at 15 months. We acknowledge the truncated lifespan is a limitation in this study. However, it was challenging to keep vehicle-treated mdx mice alive to 15 months as many developed tumors requiring euthanasia. Tumor development and shortened lifespan are afflictions that have been previously ascribed to mdx mice. 30 Since no functional cardiac impairment was observed, it was not surprising that nicorandil administration appeared to have no effect.
Comparative Summary of Findings With Respect to Short-Term Nicorandil Treatment Versus Long-Term Nicorandil Treatment in the DMD Heart.a

a A previous study in our lab examined the cardioprotective effects of short-term nicorandil administration using a sub-acute cardiac injury model with mdx mice. The current study uses the Natural History Model of DMD cardiomyopathy of mdx mice, Theoretically allowing mice to naturally develop cardiomyopathy by age 15 months, with routine, long-term administration of nicorandil throughout the life span. For ease of comparison of the findings from these 2 studies, this table summarizes the major findings of each.
Impaired skeletal muscle function in mdx mice was evident in diaphragm and treadmill analyses, but open field testing and grip strength indicated that mdx muscle function was comparable to WT. Phenotypic variability in mdx mouse behavioral analyses is a weakness of this model, as performance impairment has ranged from mild to severe. 6,69 -71 Additionally, the background of the mdx mouse represents an additional factor which introduces phenotypic variability. mdx mice with the C57Bl background often have a milder phenotype than other backgrounds, which may provide additional explanation for the lack of severity in our 15 month mdx mice colony. 72 Nicorandil did not significantly alter skeletal muscle performance in mice. Interestingly, nicorandil treatment did prolong survival, suggesting it may have had beneficial effects that were not measured in these functional assays.
Mechanistic studies of nicorandil and DMD have revealed cardioprotective benefits in in vitro and in vivo models of DMD cardiomyopathy. 14,15 Nicorandil is established as a drug that protects against cardiac injury through enhancing NO signaling, stimulating antioxidant activity and improving mitochondrial function. 20,73 Here, the antioxidant capacity of 15 month mdx hearts compared to WT was impaired, as evidenced by decreased SOD2 levels (Table 3). 47 Levels of SOD2 were unchanged by nicorandil treatment. In contrast, prior work showed nicorandil stimulated antioxidant levels of SOD2 in young mdx hearts (Table 3). 15 Further differences between the effects of nicorandil in the young mdx heart vs. the old mdx heart were evidenced by pro-oxidant NOX4 levels. Here, NOX4 levels in 15 month mdx hearts were unaltered by nicorandil, whereas nicorandil diminished NOX4 levels in young, injured mdx hearts (Table 3). 15 We initially hypothesized that NOX4 levels would be higher in mdx vs. WT, as increased activity of NADPH oxidases has previously been reported as a contributor to oxidative stress injury in DMD. 74,75 The discordant findings in the natural history model may be explained by the lack of cardiac dysfunction in this model. The sub-acute injury model induced greater cardiac injury and impaired cardiac function in contrast to the mild cardiac defect observed here in the 15 month mdx mice. mdx mice traditionally do not develop as severe of cardiac disease compared to DMD patients. 58 This was a major limitation in our study. While others have utilized additional strains of mdx mice to assess cardiomyopathy and treatment responsiveness, this project serves an important role by investigating a cardiac therapy in a well-studied DMD model.
Analyses in this study provide additional evidence that while natural history studies in DMD are important for understanding disease processes, the mdx mouse is not ideal model for investigating cardiac therapies since these mice do not sufficiently recapitulate human cardiomyopathy. We acknowledge that an alternative conclusion is that nicorandil may not be cardioprotective in DMD long-term, but we were unable to confirm or rule out this possibility due to the lack of cardiac dysfunction at 15 months. Future work investigating cardiac interventions for DMD will benefit from a combination of in vitro models for mechanistic studies and in vivo models comparable to human disease phenotypes in order to ascertain physiologic benefits of these treatments.
Methods
Animal Experiments
All animal experiments and procedures were approved by the Institutional Animal Use and Care Committee (IACUC) at the Medical College of Wisconsin (Milwaukee, WI), protocol AUA00003547. Female C57BL/10ScSn-Dmd mdx /J (mdx) or female WT C57BL/10ScSnJ mice were obtained from the Jackson Laboratory at age 11 weeks. Female mdx mice were selected for this study as they have previously been shown to develop a more severe cardiomyopathy in comparison to males. 5,66,76 Nicorandil (Selleck Chem) was given to select groups via drinking water at 6 mg/kg, daily. Nicorandil administration began at age 3 months. Experimental groups each had N = 15 and were as follows: Vehicle-treated mdx, nicorandil-treated mdx and vehicle-treated WT. Vehicle controls groups received ddH2O only. Euthanasia was performed by cervical dislocation with animals anesthetized under 2.5% isoflurane.
Study Design
Daily nicorandil or vehicle treatment began at age 3 months. All animals were subject to echocardiography and behavioral testing. Animal weights were taken 9 months, 12 months, and 15 months of age, and used for data normalization where appropriate. Behavioral testing took place at the Medical College of Wisconsin, Neuroscience Research Center Rodent Behavior Core (Milwaukee, WI). The testing procedure carried out at 9, 12 or 15 months of age was as follows: echocardiography, 4 days of rest, open field testing, 2 days of rest, grip strength testing, 2 days of rest, and treadmill testing. At the end of the 15-month protocol, hearts were harvested for protein analysis. All data was collected by researcher number 1 who assigned random numbers/letters assigned to each animal. Data was recorded under these blinded labels. All data analysis by researcher number 2, who was blinded to which animal was in which group, until all data was analyzed.
Echocardiographic Analysis of Hearts and Diaphragms
All echocardiography and measurements were performed by the Small Animal Echocardiography Core at the Medical College of Wisconsin (Milwaukee, WI). Animals were anesthetized with 2.5% isoflurane for these procedures. Measurements were taken using a Vivid iq Ultrasound System (General Electric) using 11 MHz M12-L linear array transducer. Imaging was performed as previously described. 15 The following measurements were taken: interventricular septal end diastole (IVSD), left ventricular internal diameter end diastole (LVIDd), left ventricular posterior wall end diastole (LVPWd), left ventricular internal diameter end systole (LVIDs), end diastolic volume (EDV), end systolic volume (ESV), % ejection fraction (%EF), % fractional shortening (%FS), stroke volume (SV), left ventricular mass ASE (LVD Mass ASE) and heart rate (HR).
Diaphragm measurements were taken by ultrasound sonography, adapted from a previously established protocol. 29 Briefly, the transducer was placed transversely in the subxiphoid region and angled anteriorly visualize the diaphragm at the level at which the inferior vena cava (IVC) crosses. An M-mode image was taken of the diaphragm in the region directly adjacent to the IVC during normal respiration. Using the M-mode images, respiratory rate (RR) was calculated. Parameters measured to collectively assess diaphragm function included: diaphragm excursion distance (DED), duration of inspiration (DI), velocity of inspiration (VI), duration or expiration (DE) and velocity of expiration (VE). Three consecutive breaths were analyzed for all measurements.
Multiple cardiac cycles and respirations were recorded for each ultrasound. During analysis, 3 consecutive cardiac cycles and 3 consecutive respirations were averaged for each measurement. Analyses of cardiac and respiration cycles was performed using EchoPAC with Q analysis software (General Electric).
Histology and Analysis
For histological analysis, hearts were collected at 15 months, frozen and embedded in OCT (Thermo Fisher) for sectioning. Hearts were sectioned into 8 μM thick sections for trichrome staining with Masson’s Trichrome stain (Newcomer Supply). Images were taken by the CRI Imaging Core at the Medical College of Wisconsin using a NanoZoomer HT 2.0 (Hamamatsu) at 40X magnification. Scanned images of cardiac sections were analyzed with Metamorph Software v. 7.8.0.0 (Molecular Devices) for percent fibrotic area vs. total area. Three images were analyzed per section, per animal. Values for fibrotic area were averaged per section, then per animal, then per experimental group.
Open Field Testing
The open field testing protocol was adapted from TREAT-NMD neuromuscular network standard operating procedures for DMD animal models. 77 Mice were place in a small, circular open field container with a 5” radius, 10” diameter, and 12” height. Recordings of the open field chambers were taken for 10 minutes using USB cameras. Animal movement was recorded using USB cameras. Image analysis was performed with ANY-Maze Software (Stoelting). The following measurements were taken: distance (m), mean speed (m/s), max speed (m/s), time mobile (s), time immobile (s), mobile episodes and immobile episodes.
Grip Strength Testing
Grip strength was performed using the TREAT-NMD grip strength open field standard operating procedure. 78 Measurements of forelimb and whole limb strength were taken with the Grip Strength Meter (Columbus Instruments) as previously described. 79 Briefly, mice were allowed to grip the grasping bar and were gently pulled horizontally until the grasp was broken. Measurements were discarded if all the required paws were not initially on the grip strength apparatus or if the mouse left the bars without resistance. The grip strength meter generated the peak force in the forelimb and whole limb trials. Measurements were repeated in triplicate, with breaks allowed between forelimb and whole limb testing. Measurements were averaged for each test, resulting in the maximum strength (g) for each animal. Average maximum strength was normalized to each animal’s most recent reported weight.
Treadmill Assays
Treadmill assays were adapted from the TREAT-NMD protocol and from procedures previously established in our lab designed for studying the mdx dystrophic mice 79,80 and carried out using a six-lane motorized treadmill (Columbus instruments). Mice were properly acclimated to the treadmill prior to the experiment. All mice were induced to run at increasing speeds starting at a rate of 5m/min followed by a speed increase of 1m/min each until exhaustion. The total time and distance run by each animal was recorded until exhaustion. Measurements taken included average distance traveled (m) and average duration traveled (s).
Western Blotting
Tissue was ground using a mortar and pestle and homogenized with a Dounce homogenizer as previously described in study by Sullivan et al. 15 Briefly, protein was isolated using RIPA buffer (Thermo Fisher) and Halt Protease and Phosphatase inhibitor (Thermo Fisher). Protein was quantified by BCA assay (Thermo Fisher). On gels, 20 ug protein was loaded in each lane. Primary antibodies used were NOX4 (1:300, Abcam ab133303), SOD2 (1:5000, Abcam ab13533) and HRP-conjugated vinculin (1:5000, CST #18799). The secondary antibody used was anti-rabbit HRP-conjugate (1:5000, Jackson ImmunoResearch), followed by visualization with Clarity Western ECL Substrate (GE Life Sciences). Images of blots were taken on ChemiDoc MP System (Bio-Rad) and densitometric analysis was performed using Image Lab Software (Bio-Rad). Vinculin served as a loading control for normalization. Gels were run in singlet with biological replicates.
Statistical Analysis
All animals were included in experimental analyses. Animals were randomized per cage. Animals were housed in groups of 4 per cage, according to their disease or treatment status. Performance of statistical analyses and graphs were generated in GraphPad Prism Software v. 9.1.1 (GraphPad). All data represent mean ± SEM. Statistical analyses including unpaired Student t-test one-way and two-way ANOVA were performed where appropriate and are denoted in figure legends. Kaplan-Meier survival curve analysis was performed in GraphPad. Controls for comparisons in all experiments included WT mice and vehicle treated mdx mice. For statistically rigorous experiments, N = 15 animals were in each group.
Study Limitations
A limitation of this study was the spontaneous, premature death that occurred in several mdx mice in both treatment groups. Numerous animals developed tumors requiring euthanasia, and therefore group sizes were smaller (as noted in tables) at later time points. This may have had an effect on the measurements of heart and diaphragm function to have fewer animals in the mdx groups. Additionally, the lack of cardiac dysfunction in the mdx mice compared to WT limits our ability to conclude meaningful effects of nicorandil on the DMD disease-specific cardiac defects. The lack of cardiac defect may be due to the age of the mice at terminal studies, as more advanced age may have been required for the animals to manifest cardiomyopathy. 60
Supplemental Material
Supplemental Material, sj-docx-1-cpt-10.1177_10742484221088655 - A Long-Term Study Evaluating the Effects of Nicorandil Treatment on Duchenne Muscular Dystrophy-Associated Cardiomyopathy in mdx Mice
Supplemental Material, sj-docx-1-cpt-10.1177_10742484221088655 for A Long-Term Study Evaluating the Effects of Nicorandil Treatment on Duchenne Muscular Dystrophy-Associated Cardiomyopathy in mdx Mice by Melanie Gartz, Margaret Haberman, Mariah J. Prom, Margaret J. Beatka, Jennifer L. Strande and Michael W. Lawlor in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Authors’ Note
All work in this manuscript was performed at the Medical College of Wisconsin, Milwaukee, WI. All data are available upon reasonable request. All animal experiments and procedures were approved by the Institutional Animal Use and Care Committee (IACUC) at the Medical College of Wisconsin (Milwaukee, WI), protocol AUA00003547.
Acknowledgments
The authors would like to thank Leanne Harmann and Kaitlin Duskin of the Small Animal Echocardiography Core at the Medical College of Wisconsin for performing echocardiography and analysis. They also thank Suresh Kumar of the Imaging Core at the Medical College of Wisconsin for imaging histology sections.
Author Contributions
MG wrote the manuscript, performed statistical analysis, performed histological analysis, and interpreted results from experiments. MH performed all mouse experiments and analyzed data. MJP and MJB performed and analyzed all western blots. JLS conceived this project, oversaw all mouse experiments, and provided guidance on data analysis. MWL oversaw western blot analysis and histological analysis and revised this manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MWL is or has recently been a member of advisory boards for Solid Biosciences, Taysha Therapeutics, and Astellas Gene Therapies (formerly Audentes Therapeutics). MWL is also a consultant for Astellas Gene Therapies (formerly Audentes Therapeutics), Encoded Therapeutics, Modis Therapeutics, Lacerta Therapeutics, Dynacure, AGADA Biosciences, Affinia Therapeutics, and Biomarin. MWL receives research support from Astellas Gene Therapies, Solid Biosciences, Kate Therapeutics, and Prothelia.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the National Institutes of Health grant awarded to JLS and MWL (NIH R01HL134932). The above funding bodies had no role in design and execution of this project.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.