
Abstract
Background:
Dystrophin-deficient cardiomyopathy is a growing clinical problem without targeted treatments. We investigated whether nicorandil promotes cardioprotection in human dystrophin-deficient induced pluripotent stem cell (iPSC)-derived cardiomyocytes and the muscular dystrophy mdx mouse heart.
Methods and Results:
Dystrophin-deficient iPSC-derived cardiomyocytes had decreased levels of endothelial nitric oxide synthase and neuronal nitric oxide synthase. The dystrophin-deficient cardiomyocytes had increased cell injury and death after 2 hours of stress and recovery. This was associated with increased levels of reactive oxygen species and dissipation of the mitochondrial membrane potential. Nicorandil pretreatment was able to abolish these stress-induced changes through a mechanism that involved the nitric oxide–cyclic guanosine monophosphate pathway and mitochondrial adenosine triphosphate-sensitive potassium channels. The increased reactive oxygen species levels in the dystrophin-deficient cardiomyocytes were associated with diminished expression of select antioxidant genes and increased activity of xanthine oxidase. Furthermore, nicorandil was found to improve the restoration of cardiac function after ischemia and reperfusion in the isolated mdx mouse heart.
Conclusion:
Nicorandil protects against stress-induced cell death in dystrophin-deficient cardiomyocytes and preserves cardiac function in the mdx mouse heart subjected to ischemia and reperfusion injury. This suggests a potential therapeutic role for nicorandil in dystrophin-deficient cardiomyopathy.
Introduction
Cardiomyopathy is a devastating and inescapable consequence for patients with Duchenne or Becker muscular dystrophy. Dystrophin gene mutations cause dystrophin deficiency either through the complete absence of dystrophin or the aberrant expression of an internally truncated dystrophin protein in cardiac and skeletal muscle. The first manifestation of dystrophin gene mutations is typically progressive skeletal muscle weakness starting within the first decade of life, while cardiac involvement develops in the second decade. The addition of steroid treatment and advances in respiratory care have prolonged the lives of these patients; accordingly, cardiac disease has become a leading cause of death. 1,2
Dystrophin deficiency in the mouse mdx, a widely studied animal model of Duchenne muscular dystrophy, is associated with several cellular abnormalities including increased calcium influx, disruption of the nitric oxide (NO)–cyclic guanosine monophosphate (cGMP) signaling, increased generation of reactive oxygen species (ROS), and mitochondrial dysfunction within the myocytes. 3 –6 These deleterious consequences combine to promote cell death. Using one compound that modulates several of these pathological mechanisms may be of value in protecting against dystrophin-deficient cardiomyopathy in the clinical setting. Nicorandil (2-[(pyridin-3-ylcarbonyl)amino] ethyl nitrate) may be one such agent. Nicorandil acts as an NO donor and a KATP channel opener. 7 –9 It also acts as an antioxidant by increasing superoxide dismutase 2 (SOD2) and decreasing xanthine oxidase activity. 10 Through these pleiotropic mechanisms, it activates the NO-cGMP pathway, 11,12 decreases oxidative stress, preserves mitochondrial function, and decreases both hypoxia-induced and oxidative stress-induced apoptosis in neonatal rat ventricular myocytes. 12 –15 Even though similar mechanisms play a detrimental role in the dystrophin-deficient cardiomyocyte, no studies have examined whether nicorandil may be beneficial for dystrophin-deficient cardiomyopathy.
We and others have previously shown induced pluripotent stem cell (iPSC) derived from patients with muscular dystrophy (Dys-iPSCs) and differentiated into cardiomyocytes (iPSC-CMs or iCMs) is a useful model to study mechanisms and treatment approaches for dystrophin-deficient cardiomyopathy. 16,17 The purpose of this study was to determine whether nicorandil mitigates stress-induced injury in the dystrophin-deficient human iPSC-derived cardiomyocytes and restores cardiac function in the dystrophin-deficient mdx mouse heart.
Methods and Materials
Animals and Reagents
Male C57Bl/6 and C57BL/10ScSn-mdx/J (mdx) mice at 11 to 12 weeks of age (Jackson Laboratories, Bar Harbor, Maine) used in this study received humane care in compliance with the Guide for the Care and Use of Laboratory Animals formulated by the National Research Council (2015) and approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin. Nicorandil was obtained from SelleckChem (Houston, Texas). All other reagents were obtained from Sigma (St Louis, Missouri) unless otherwise specified.
Human iPSC Culture and Myocyte Differentiation
The use of human iPSC lines was approved by the institutional review board of the Medical College of Wisconsin (PRO00015971). The iPSC lines used were previously generated from male subjects who were diagnosed with Duchenne muscular dystrophy (Dys) or nonaffected (N) male subjects. 18,19 The Dys1 line contains an out-of-frame dystrophin gene deletion of exons 3 to 6 (Systems Biosciences, Mountain View, California), and the Dys2 line contains an in-frame deletion of exons 45 to 53 (Coriell Institute, Camden, New Jersey). 19 The N-iPSC line was a generous gift from Stephen Duncan. 18 All iPSC lines were used between passages 40 and 80 and maintained on Matrigel (BD Biosciences, San Jose, California) in mTeSR-1 media (Stem Cell Technologies, British Columbia, Canada) in a 5% CO2 incubator with atmospheric (20%) O2 at 37°C. The pluripotent and undifferentiated iPSC state was determined periodically (every 2-3 months) by reverse transcription polymerase chain reaction (RT-PCR) and immunofluorescence staining of pluripotency markers such as Oct3/4 and absence of endoderm, mesoderm, and cardiac lineage markers such as Gata-1, NKX2-5, α-actinin, MYH6, MYH7, and MLC-2v (Supplementary Figure S1). Induced pluripotent stem cell differentiation into the cardiac lineage was achieved by adapting published protocols. 20,21 The N-iPSCs and Dys2-iPSCs were differentiated using a small molecule protocol, 21 whereas the Dys1-iPSCs were differentiated using the Matrigel sandwich protocol. 20 Differentiated cells were maintained in cardiomyocyte maintenance media (Roswell Park Memorial Institute [RPMI]/B27 with 11.11 mmol/L glucose; Life Technologies, Grand Island, New York). Induced cardiomyocytes were tagged for live cell imaging by transduction with a lentiviral plasmid encoding for enhanced green fluorescent protein (eGFP; multiplicity of infection (MOI): 5.67 × 109 IU/mL) under the control of the cardiac-specific sodium–calcium exchanger 1 (NCX-1) promoter 22 as previously described. 23 For all experiments, 35 ± 5-day-old contracting iCMs that were confirmed to be >70% cardiomyocytes by cardiac troponin T staining or eGFP-positive fluorescence were used for further analysis.
Skeletal muscle differentiation was performed according to a previously published protocol. 24 Induced pluripotent stem cell-derived myotubes were cultured for up to 6 weeks in Dulbecco modified eagle medium (DMEM)/B27.
Reverse Transcription Polymerase Chain Reaction and Quantitative Polymerase Chain Reaction
Total RNA samples were isolated using the Qiagen RNeasy mini kit (Valencia, California). Complementary DNAs (cDNAs) were synthesized using the iScript cDNA Synthesis kit (Bio-Rad Laboratories, Hercules, California). The forward and reverse primer sequences used in RT-PCR reactions are dystrophin exon 3 to 5 (sense 5′-GGCAAAAACTGCCAAAAGAA-3′ and antisense 5′-GACCTGCCAGTGGAGGATTA-3′) and exon 47 to 48 (sense 5′-CCATAAGCCCAGAAGAGCAA-3′ and antisense 5′-AAGCTGCCCAAGGTCTTTTA-3′). The amplification conditions were 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 30 seconds for 35 cycles, and 72°C for 7 minutes for 1 cycle. DNA electrophoresis was conducted on a 2% agarose gel.
Quantitative real-time PCR (qPCR) was performed on the CFX96 Thermal Cycler (Bio-Rad) with iTaq Universal SYBR Green supermix (Bio-Rad). Nitric oxide synthase isoforms 1 to 3 and SOD2 gene expression were analyzed using the prevalidated QuantiTect primer assay by Qiagen (QT00043372, QT00068740, QT00089033, and QT01008693) and assayed in triplicate. Glyceraldehyde 3-phosphate dehydrogenase (primers: sense 5′-GTGGACCTGACCTGCCGTCT-3′ and antisense 5′-GGAGGAGTGGGTGTCGCTGT-3′) was used for normalization. The cDNA templates of 25 ng were used for each amplification reaction. The double ΔCt method was used to quantify relative gene expression. 25
Single-Cell Dissociation
Single cells were dissociated using 0.05% Trypsin-EDTA (Invitrogen, Carlsbad, California) for 5 minutes. Trypsin was inactivated with DMEM + 10% fetal calf serum. Matrigel-coated coverslips (BD Biosciences) were seeded at a density of 12 000 cells/cm2.
Immunofluorescence
Immunostaining was performed using anticardiac troponin T (Abcam, Cambridge, Massechusetts) and antidystrophin (NCL-DYSB, against dystrophin protein epitope amino acids 321-494 encoded by exons 10-12; Leica Biosystems, Buffalo Grove, Illinois) along with labeled secondary antibodies (Life Technologies). Cells were counterstained with 4’,6-diamidino-2-phenylindole or Hoechst nuclear stains. Images were captured with a confocal microscope (Nikon A1-R; Nikon Instruments, Melville, New York).
Stress and Recovery-Induced Injury
To induce a stress response, cells were subjected to 100 μmol/L H2O2 and 10 mmol/L deoxyglucose in RPMI (without glucose) for 2 hours and recovery in cardiomyocyte maintenance media (RPMI/B27 with 11.11 mmol/L glucose) for 4 hours. For control experiments, cells were exposed to RPMI (with 11.11 mmol/L glucose) for 2 hours followed by 4 hours in cardiomyocyte maintenance media (RPMI/B27 with 11.11 mmol/L glucose). The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed after 24-hour recovery. All other assays were performed after 4 hours of recovery.
Reactive Oxygen Species and Mitochondrial Membrane Potential Assays
Dihydroethidium (DHE, 10 μmol/L) or tetramethylrhodamine ethyl ester (TMRE; 50 nmol/L) was used to detect ROS or mitochondrial membrane potential (ΔΨm) in eGFP-labeled iCMs. Cells were treated for 20 minutes with DHE or TMRE after the 4-hour recovery period. Images were taken immediately after a 5-minute wash period using confocal microscopy with a ×40 objective. The fluorescent intensity of the ethidium derivative or TMRE was detected by a laser excitation/emission (ex/em) 518/605 nm or ex/em 540/595 nm, respectively. The changes in membrane potential were monitored by calculating relative TMRE fluorescence. Five regions of interests (ROIs) were selected from the nucleus (DHE) or mitochondria (TMRE) of eGFP-positive iCMs and measured for mean fluorescent intensity (ImageJ, version 1.48v, Java 1.6.0_65; National Institutes of Health, Bethesda, Maryland). Imaging conditions such as gain levels, frames per second, and aperture size were held constant.
In selected experiments, iCMs were pretreated with nicorandil (100 μmol/L for 24 hours), with or without 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10 μmol/L, an inhibitor of soluble guanylyl cyclase) and 5-hydroxydecanoate (5-HD, 500 μmol/L, a mitochondrial KATP channel antagonist). These inhibitors were added 15 minutes prior to nicorandil.
Measuring Mitochondrial Permeability Transition Pore Opening
To induce mitochondrial permeability transition pore (mPTP) opening, cells were loaded with TMRE for 25 minutes at room temperature. On laser illumination, TMRE generates ROS within the mitochondria leading to mPTP opening and visualized by loss of the TMRE fluorescence. Time required to induce mPTP opening was determined from ΔΨm recordings. The peak signal value over the recorded region (50 μm2) was normalized as 100% and the lowest value as 0%. After normalization, the time required for a 50% decrease in signal was calculated and denoted as t.
Hydrogen Peroxide Measurements
Cardiomyocyte-specific cytosolic or mitochondrial hydrogen peroxide was detected using a compartment-specific fluorescent genetic probe directed to either the cytosol (HyPer-Cyto) or the mitochondria (HyPer-Mito). The probe was generated by placing the HyPer-Cyto or HyPer-Mito cassette (Axxora, LLC, Farmingdale, New York) downstream from the NCX-1 promoter within a lentiviral plasmid. Five days postdissociation, iCMs were infected with NCX-1-HyPer-Cyto or NCX-1-HyPer-Mito lentivirus vectors (MOI: 7.35 × 108 or 2.24 × 109 IU/mL, respectively). For hydrogen peroxide measurements, cells were exposed to the control or stress and recovery (S/R) conditions and stained with Hoechst for nuclear staining. Fluorescence was observed using confocal microscopy and analyzed with ImageJ software as described above.
Cell Injury and Death Assays
Cell injury was assessed by measuring lactate dehydrogenase (LDH) release into the supernatants using a colorimetric cytotoxicity assay kit according to the manufacturer’s directions (Roche, Indianapolis, Indiana). The percentage LDH release was normalized to a no-stress vehicle control group for each cell line.
Cell death was evaluated using a TUNEL assay (Roche) per the manufacturer’s instruction. Cells were counterstained with Hoechst. Negative controls were set up per the manufacturer’s instructions, and DNAse-treated iCMs were used as a positive control. Coverslips were mounted, and cells were analyzed by fluorescent microscopy. A series of 10 images were taken from randomly selected areas on the coverslip. The TUNEL (+) nuclei in each image were quantified and compared against Hoechst-stained nuclei to give a percentage of TUNEL (+) cells.
Isolated Heart Perfusion
Mice were anesthetized by intraperitoneal administration of sodium pentobarbital (80 mg/kg), and once the pedal reflex was lost, the hearts were excised. Isolated hearts were perfused retrograde through the aorta with a modified Krebs buffer and instrumented as previously described. 26 Briefly, hearts were perfused for 30 minutes for stabilization, and baseline left ventricular (LV) contraction and coronary flow were recorded. Hearts were then subjected to 30 minutes of global ischemia and 120 minutes of reperfusion. In the treatment group, 20 μmol/L nicorandil was administered for 20 minutes prior to ischemia. This dose has previously been shown to be cardioprotective in the isolated mouse heart. 27 Coronary flow was monitored by an in-line flow probe connected to a flow meter (Transonics Systems Inc, Ithaca, New York). A saline-filled latex balloon connected to a pressure transducer was inserted into the LV, and baseline end-diastolic pressure was set at 5 to 10 mm Hg. Left ventricular developed pressure (LVDP), maximum rate of increase in LVDP (+dP/dt), and maximum rate of decrease in LVDP (−dP/dt) at baseline and 30, 60, and 120 minutes after reperfusion were determined. Wild-type (WT) and mdx mice were randomly assigned to either vehicle or nicorandil groups (n = 8 mice/group). Wild-type sham isolated hearts that were perfused but not subjected to global hypoxia were also included.
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). Each experimental group consisted of iCMs from at least 3 separate differentiations performed in technical triplicates; n indicates the number of independent experiments. For the statistical analysis, SigmaPlot software (version 11, SigmaPlot; Systat Software, Inc, Richmond, California) was used. Statistical comparisons between the 3 groups for each condition were determined by 1-way analysis of variance followed by Student-Newman-Keuls post hoc test to analyze group differences. The Student t test was used when appropriate. P ≤ .05 were considered statistically significant.
Results
Characterization of iPSCs-Derived Cardiomyocytes
Dystrophin transcripts were expressed in the all iCM cultures; however, transcripts containing exons 3 to 5 or exons 47 to 48 were not detected in Dys1-iCMs or Dys2-iCMs, thereby confirming the specific deletion mutation (Figure 1A). Immunostaining revealed dystrophin in N-iCMs but not in Dys1-iCMs (Figure 1B, left and center panels). The Dys2-iCMs, containing an internally truncated dystrophin, had an overall weaker staining for dystrophin when compared to the N-iCMs (Figure 1B, right panel). Induced pluripotent stem cell-derived cardiomyocytes were either identified by cardiac troponin T staining or eGFP-positive fluorescence (Figure 1B, middle and lower panels). Despite using different cardiac differentiation protocols, once differentiated, the 3 lines had similar upregulation of cardiac-specific markers such as by immunofluorescence and RT-PCR (Supplementary Figure S2).

Assessment of dystrophin exon deletions and dystrophin expression in induced pluripotent stem cell (iPSC)-derived cardiomyocytes. Nonaffected (N) iPSCs and 2 unrelated iPSC lines generated from subjects with Duchenne muscular dystrophy (Dys1-iPSC, containing an exon 3-6 deletion and Dys2-iPSC, containing an exon 45-53 deletion) were differentiated into cardiomyocytes (iCMs) and assessed for specific dystrophin exon deletions by reverse transcription polymerase chain reaction (RT-PCR) and dystrophin protein expression by immunostaining. A, Polymerase chain reaction (PCR) amplification confirms exons 3 to 5 and 47 to 48 in N-iCMs. In Dys1-iCMs, exons 3 to 5 are not amplified, but exons 47 to 48 are present. In the Dys2-iCMs, exons 3 to 5 are amplified but not exons 47 to 48, thus confirming the expected mutations in the Dys-iCMs. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was a positive control. B, Representative immunostaining of iCMs using antibodies against dystrophin (upper panel) and costaining of dystrophin and cardiac troponin T (cTNT; middle panel). Dystrophin is detected in troponin-positive N-iCMs but not in Dys1-iCMs. The Dys2-iCMs display weaker dystrophin staining in troponin-positive cells. The expression of the cardiac-specific eGFP to label cardiomyocytes is shown in the lower panel. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI).
Dystrophin deficiency is associated with nitric oxide synthase (NOS) mislocalization and/or deficiency in myocytes. 28,29 Neuronal NOS (nNOS) interacts with dystrophin-associated glycoprotein complex at the sarcolemma in skeletal myocytes, and this association is lost in dystrophin deficiency. 28 However, in cardiomyocytes isolated from the mdx mouse, nNOS does not colocalize with dystrophin 30,31 but instead colocalizes with the ryanodine receptor 2 (RyR2) at the sarcoplasmic reticulum. 32 Endothelial NOS localizes to the Golgi complex and the sarcolemma caveolae. 33 Immunostaining for dystrophin and nNOS in iCMs did not reveal any overlapping staining at the sarcolemma (Figure 2A). Neuronal NOS is localized to the RyR2 in N-iCMs (Supplementary Figure S3A). Immunostaining for dystrophin and endothelial NOS (eNOS) shows that both proteins localize to the sarcolemma (Figure 2B), and the costaining pattern suggests that dystrophin and eNOS are in close proximity in the N-iCMs (left panels). Dystrophin is undetectable in the Dys1-iCMs, although eNOS is still present. In the Dys2-iCM line, which encodes for an internally truncated form of dystrophin, the overlapping staining pattern of eNOS and dystrophin at the sarcolemma is maintained (Figure 2B, right panels). In the iCMs, eNOS colocalizes with caveolin 1, a caveolae marker, whether dystrophin is present (Supplementary Figure S3B).
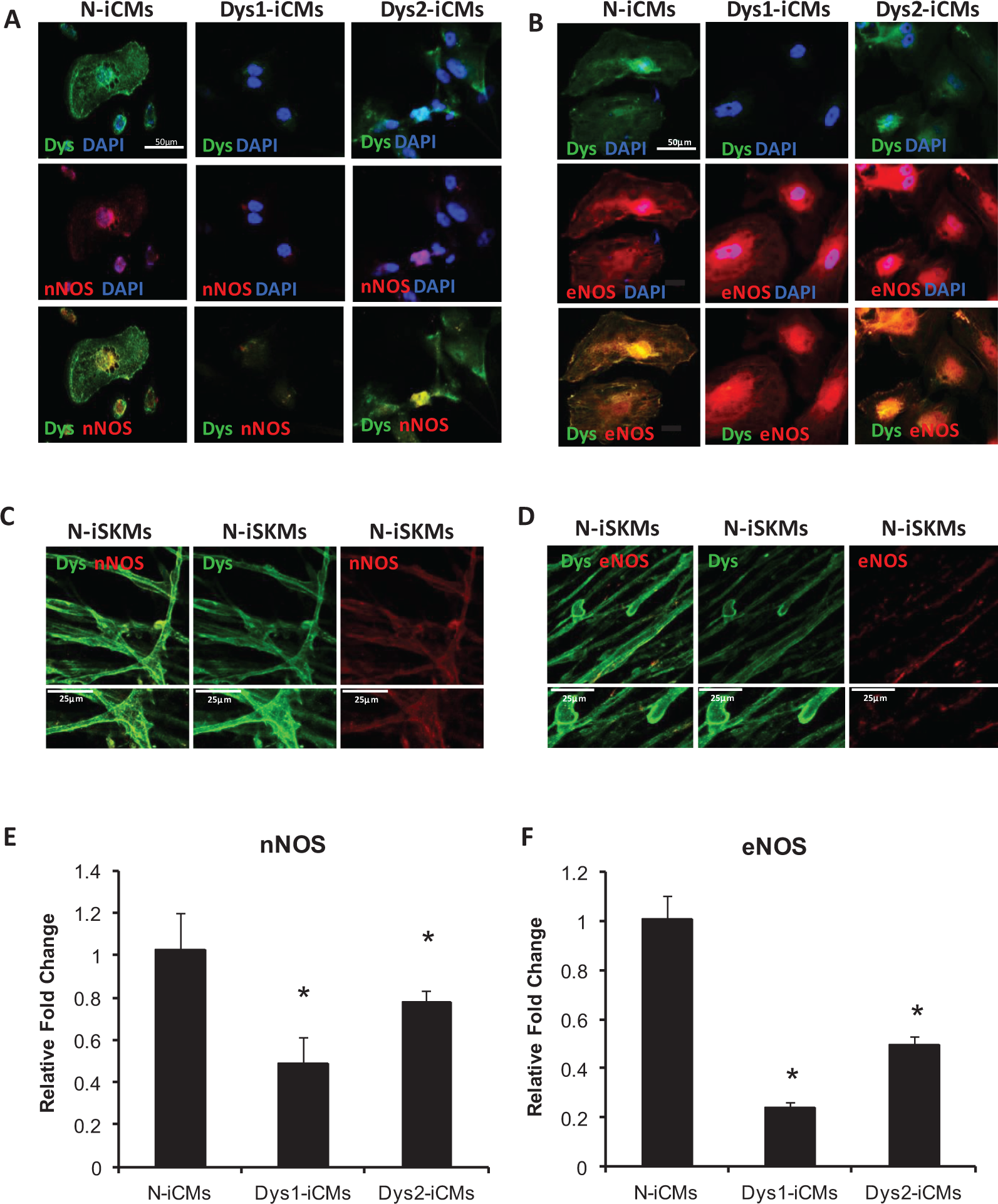
Nitric oxide synthase protein localization and gene expression levels in nonaffected and dystrophin-deficient induced pluripotent stem cell (iPSC)-derived myocytes. Nonaffected (N)-iPSCs and 2 dystrophin-deficient (Dys1 and Dys2) iPSC lines were differentiated into cardiomyocytes (iCMs) and assessed for dystrophin and either neuronal nitric oxide synthase (nNOS) or endothelial NOS (eNOS) cellular location by confocal microscopy. A, Dystrophin (upper panels) is distributed to the sarcolemma in N-iCMs and to a smaller extent in Dys2-iCMs, whereas nNOS (middle panels) is detected in the cytosol in all groups. The merged images (lower panels) show that dystrophin and nNOS staining do not overlap at the sarcolemma in the iCMs. Scale bar = 50 μmol/L. B, Dystrophin (upper panels) is localized to the sarcolemma along with eNOS (middle panels) in N-iCMs. The close proximity of dystrophin and eNOS is confirmed in the merged lower panel. The proximity of eNOS with dystrophin cannot be assessed in the Dys1-iCM line, which does not express dystrophin, however, in the Dys2-iCMs that express an internally truncated form of dystrophin, eNOS and dystrophin are found in close proximity at the sarcolemma. Scale bar = 50 μmol/L. C and D, In N-iPSC-derived skeletal myocytes, dystrophin is colocalized with nNOS at the sarcolemma (scale bar = 25 μmol/L; C) but dystrophin does not colocalized with eNOS (scale bar = 25 μmol/L; D). E and F, Quantitative real-time PCR (qPCR) reveals that nNOS (E) and eNOS (F) messenger RNA (mRNA) levels are decreased in Dys-iCMs when compared to N-iCMs. Data are mean ± standard error of the mean (SEM) of values from 3 to 6 pooled independent cardiac differentiations, run in triplicate. *P ≤ .05 versus N-iCMs.
For comparison, we used skeletal myocytes derived from N-iPSCs. In these cells, immunostaining for nNOS and dystrophin overlaps at the sarcolemma (Figure 2C), whereas eNOS does not (Figure 2D).
We were not able to detect NO in the iCMs using different NO sensors, including 4,5-diaminofluorescein diacetate and DAXJ2, which have a lower limit of detection of 5 nmol/L; therefore, we assessed gene expression levels for nNOS, eNOS, and inducible NOS (iNOS) using qPCR. The messenger RNA (mRNA) levels of nNOS and eNOS were decreased in Dys1-iCMs and Dys2-iCMs compared to N-iCMs (Figure 2E and F). There was no significant change in iNOS mRNA levels (Supplementary Figure S4).
Effects of Nicorandil on S/R-Induced Injury of iPSC-Derived Cardiomyocytes
As shown in Figure 3A, pretreatment with nicorandil (0.01-100 μmol/L) elicited a concentration-dependent protection against S/R-induced injury in N-iCMs as detected by LDH release into the supernatant. In addition, nicorandil abolished S/R-induced increase in ROS levels in a concentration-dependent manner (Figure 3B) as detected by an increase in DHE fluorescence. The maximal cardioprotective effects of nicorandil were observed with the 100 μmol/L concentration; therefore, this dose was selected for all other experiments.

Concentration-dependent effects of nicorandil to protect against stress and recovery (S/R)-induced cell injury and reactive oxygen species (ROS) production in nonaffected induced pluripotent stem cell (iPSC)-derived cardiomyocytes. The N-iCMs were subjected to 2 hours of oxidative stress followed by recovery. Protective effects of different concentrations of nicorandil (0.1, 1, 10, and 100 μmol/L) on cell injury and ROS production were assessed after 4 hours of recovery by measuring lactate dehydrogenase (LDH) release into the supernatant and evaluating dihydroethidium (DHE) fluorescent intensity, respectively. A, Stress induces an increase in LDH release in N-iCMs, which is mitigated by 10 and 100 μmol/L nicorandil but not 0.1 and 1 μmol/L. B, Stress induced an increase in ROS levels as indicated by DHE fluorescence, which is abolished by 10 and 100 μmol/L nicorandil but not 0.1 and 1 μmol/L. Data are mean ± standard error of the mean (SEM) of values from 3 independent experiments (n = 3/group). *P ≤ .05 versus control.
Nicorandil Protects Against S/R-Induced Cell Injury and Death After Stress in Dystrophin-Deficient iPSC-Derived Cardiomyocytes
To investigate whether nicorandil mitigates S/R-induced injury and death, we measured LDH release and examined DNA damage by TUNEL staining. Stress and recovery significantly increased LDH release in the untreated Dys-iCMs when compared to N-iCMs, which was mitigated by nicorandil (Figure 4A). Likewise, nicorandil mitigated the increase in TUNEL-positive cells after S/R when compared to untreated cells. Under control conditions, there is an increased number of TUNEL-positive Dys1-iCMs (Figure 4B), which may be due to mechanical damage during the washing steps. Furthermore, exposure to stress significantly increased the number of TUNEL-positive cells in the Dys1-iCMs and Dys2-iCMs compared to N-iCMs. Because nicorandil acts as an NO donor and a KATP channel opener, we used ODQ and 5-HD to inhibit the NO-cGMP pathway and KATP channel opening, respectively. The protective effect of nicorandil was only partially inhibited by ODQ or 5-HD and was completely abolished by coapplication of ODQ and 5-HD. Nicorandil decreased cell injury and death in an NO-cGMP and a KATP channel-dependent manner.

Nicorandil prevents stress-induced cell injury and death in dystrophin-deficient induced pluripotent stem cell (iPSC)-derived cardiomyocytes. Cells were treated for 24 hours with nicorandil with or without 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), a selective inhibitor of nitric oxide (NO)-sensitive guanylyl cyclase, and 5-hydroxydecanoate (5-HD), an inhibitor of mitoKATP channel opening prior to 2 hours of oxidative stress. Cell injury was assessed after 4 hours of recovery by measuring LDH release into the supernatant, and cell death was measured after 24 hours by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive staining. A, Stress induces an increase in LDH release in all groups but is significantly elevated in Dys1-iCMs and Dys-2 iCMs. Nicorandil is able to prevent the stress-induced injury. The ODQ and 5-HD partially reverse nicorandil’s protective effects. B, Cell death was assessed by TUNEL staining after 24 hours of recovery. The Dys1-iCMs have a higher rate of death under control conditions. Stress increases the rate of cell death in iCMs and even more so in Dys-iCMs. Nicorandil is able to prevent the increase in stress-induced death. The ODQ and 5-HD partially abolish the protective effects of nicorandil. Data are mean ± standard error of the mean (SEM) of values from 3 independent experiments (n = 3/group). *P ≤ .05 versus control, #P ≤ .05 versus N-iCMs, &P ≤ .05 versus stress, γP ≤ .05 versus stress + nicorandil (nico).
Effect of Nicorandil Intracellular ROS on Mitochondrial Integrity After Stress in Dystrophin-Deficient iPSC-Derived Cardiomyocytes
Stress and recovery increased the level of intracellular ROS detected based on the fluorescence of DHE in each group. However, S/R induced a significantly higher ROS levels in the dystrophin-deficient cardiomyocytes when compared to N-iCMs. Furthermore, the Dys1-iCM group had increased ROS levels under control conditions. Nicorandil decreased the level of intracellular ROS detected in all groups (Figure 5A). The ODQ abolished the ability of nicorandil to decrease ROS levels in N-iCMs and partially in Dys2-iCMs but not in Dys1-iCMS. 5-Hydroxydecanoate did not reverse the protective effects of nicorandil. The ODQ and 5-HD alone had no effects on ROS levels (Supplementary Figure S5A). Nicorandil decreased ROS levels partially through the NO-cGMP pathway but independent of KATP channel function, suggesting that nicorandil may have antioxidant effects independent of its action on NO-cGMP and KATP channels in dystrophin-deficient cardiomyocytes.

Nicorandil normalizes reactive oxygen species (ROS) levels and maintains the mitochondrial after stress in dystrophin-deficient induced pluripotent stem cell (iPSC)-derived cardiomyocytes. Cells were subjected to 2 hours of oxidative stress followed by 4 hours of recovery with or without 24-hour pretreatment with nicorandil and with or without 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), a selective inhibitor of nitric oxide (NO)-sensitive guanylyl cyclase, and 5-hydroxydecanoate (5-HD), an inhibitor of mitoKATP channel opening. Cells were stained with tetramethylrhodamine ethyl ester (TMRE) to assess the mitochondrial membrane potential or with dihydroethidium (DHE) to detect the production of ROS. A, Quantitative analysis of DHE fluorescence. Stress induced an increase in ROS levels, which was mitigated by pretreatment of nicorandil. The ODQ inhibits nicorandil in N-iCMs and Dys2-iCMs but not Dys1-iCMs, and 5-HD has no effect on nicorandil. B, Quantitative analysis of TMRE fluorescence. Stress decreased mitochondrial membrane potential in Dys-iCMs, and nicorandil abrogates the effects of stress. The ODQ and 5-HD abolish the protective effects of nicorandil. Data are mean ± standard error of the mean (SEM) of values from 3 independent experiments (n = 3/group). *P ≤ .05 versus control, #P ≤ .05 versus N-iCMs, &P ≤ .05 versus stress, γP ≤ .05 versus stress + nico.
Increased ROS is known to cause formation of the mPTP and depolarization of the mitochondrial membrane potential 34 –36 ; therefore, we determined whether nicorandil was able to maintain the mitochondrial membrane potential. Stress and recovery decreased the fluorescence intensity of TMRE, reflecting the depolarization of the mitochondrial membrane potential. Pretreatment with nicorandil was able to protect against the S/R-induced loss of mitochondrial membrane potential in the Dys-iCMs and increase the membrane potential in N-iCMs (Figure 5B). Both ODQ and 5-HD were able to block the protective effects of nicorandil on the mitochondrial membrane potential. Coapplication of ODQ and 5-HD had additive effects. The ODQ and 5-HD had no effect alone (Supplementary Figure S5B). Furthermore, oxidative stress causes earlier opening of the mPTP in the Dys1-iCMs and Dys2-iCMs. Pretreatment with nicorandil is able to delay mPTP opening time in both nonaffected and dystrophin-deficient iCMs (Supplementary Figure S6).
Nicorandil Limits Mitochondrial ROS Production
Nicorandil decreases ROS by increasing antioxidant gene expression such as SOD2.10 Superoxide dismutase 2 is a major antioxidant in mitochondria. Superoxide dismutase 2 expression was decreased at baseline in the Dys-iCMs when compared to the N-iCMs (Figure 6A). Superoxide dismutase 2 levels increase after 2 hours of stress and 4 hours of recovery in the N-iCMs but not in the Dys-iCMs groups (Figure 6B), suggesting that dystrophin deficiency abrogates the ability of cell to increase SOD2 expression. Nicorandil pretreatment increased SOD2 levels in the dystrophin-deficient cells (Figure 6B). Since SOD2 converts superoxide to hydrogen peroxide, we measured the levels of mitochondrial hydrogen peroxide using a genetic hydrogen peroxide sensor HyPer, directed to the mitochondria. The levels of hydrogen peroxide were significantly increased in mitochondria of Dys1-iCMs at baseline when compared to the Dys2-iCM and N-iCMs. There was a significant increase in mitochondrial hydrogen peroxide in all groups following S/R, and nicorandil prevented the increase in mitochondrial hydrogen peroxide (Figure 6C).

Nicorandil increases superoxide dismutase 2 (SOD2) expression and decreases mitochondrial reactive oxygen species (ROS) levels after stress. A, Quantitative polymerase chain reaction (PCR) analysis of SOD2 gene transcripts revealed that SOD2 expression levels are decreased in the Dys1-iCMs and Dys2-iCMs at baseline when normalized to N-iCMs. B, N-iCMs, Dys1-iCMs, and Dys2-iCMs were subjected to 2 hours of oxidative stress followed by 4 hours of recovery with or without nicorandil. The SOD2 expression levels were normalized to control conditions within each group. Stress increases SOD2 expression in N-iCMs but not Dys-iCMs. Nicorandil restores SOD2 levels after stress in the Dys-iCMs. C, The iCMs were transduced with a fluorescent probe that detects hydrogen peroxide in the mitochondria (HyPer-Mito) and subjected to control or stress and recovery (S/R) conditions. The S/R induced an increase in mitochondrial hydrogen peroxide, which was mitigated by the pretreatment with nicorandil. Data are mean ± standard error of the mean (SEM) of values from 3 independent experiments (n = 3/group). *P ≤ .05 versus control, #P ≤ .05 versus N-iCMs, &P ≤ .05 versus stress.
The finding of increased levels of hydrogen peroxide in the Dys1-iCMs was incongruent to the finding of decreased expression of SOD2 in these cells at baseline. Therefore, we postulated that the dystrophin-deficient cells may have compromised hydrogen peroxide clearance mechanisms. Considering that hydrogen peroxide is cleared by the oxidation of NADPH by the glutathione peroxidase (GPX1)/glutathione reductase (GSR) system, we evaluated gene expression levels of GPX1 and GSR, two of several genes involved in hydrogen peroxide clearance. Supplementary Figure S7 shows that there is decreased expression of GPX1 and GSR genes in the Dys-iCMs when compared to the N-iCMs.
Nicorandil Limits Cytosolic ROS Production by Xanthine Oxidase
Nicorandil has also been shown to decrease ROS by inhibiting xanthine oxidase activity. 10 Therefore, we assessed whether xanthine oxidase is contributing to the increased ROS levels in the Dys-iCMs. To do this, we pretreated the iCMs with xanthine oxidase inhibitors, allopurinol and febuxostat. Figure 7A shows that both xanthine oxidase inhibitors decrease ROS. Furthermore, we show in Figure 7B that nicorandil decreases cytosolic hydrogen peroxide production.

Xanthine oxidase inhibitors decrease stress-induced reactive oxygen species (ROS) production in induced pluripotent stem cell (iPSC)-derived cardiomyocytes, and nicorandil decreases cytosolic ROS level. A, Cells were subjected to either control (no stress) or to stress and recovery (S/R) conditions (2 hours of stress followed by 4 hours of recovery) with or without xanthine oxidase inhibitors allopurinol and febuxostat. Cells were stained with dihydroethidium (DHE) to detect ROS, and a defined area in each cardiomyocyte was quantitated for the intensity of DHE fluorescence. Allopurinol and febuxostat prevented the increase of ROS after stress. B, Cells were transduced with a fluorescent probe that detects hydrogen peroxide in the cytosol (HyPer-Cyto) and subjected to control or S/R conditions. There is increased fluorescence in the Dys1-iCMs and Dys2-iCMs after S/R compared to the N-iCMs. Nicorandil decreased cytosolic ROS levels. Data are mean ± standard error of the mean (SEM) of values from 3 independent experiments (n = 3/group). *P ≤ .05 versus control, #P ≤ .05 versus N-iCMs, &P ≤ .05 versus stress.
Nicorandil Improves Functional Recovery in Perfused mdx Hearts
The effects of nicorandil on cardiac function were examined in hearts isolated from mdx and control mice. Baseline values of LVDP, measurements of global contractility (+dP/dt), diastolic function (−dP/dt), LV end-diastolic pressure, and coronary flow were comparable among groups (Table 1). The isolated hearts were subjected to 30-minute global ischemia and 2-hour reperfusion to unmask a phenotype. The decline in LVDP, +dP/dt, and −dP/dt was significantly worse in the mdx hearts than controls. Pretreatment with nicorandil significantly increased LVDP, +dP/dt, and −dP/dt in both the WT and mdx groups when compared to the vehicle-treated groups. Nicorandil increased recovery of LVDP in the mdx and control hearts (Figure 8).
Hemodynamics in the Isolated Perfused Hearts.

Abbreviations: +dP/dt; −dP/dt; LV, left ventricle; WT, wild type.
aP < .05 versus WT + vehicle.
bP < .05 versus sham.
cP < .05 versus WT + nicorandil.
dP < .05 versus mdx + vehicle.
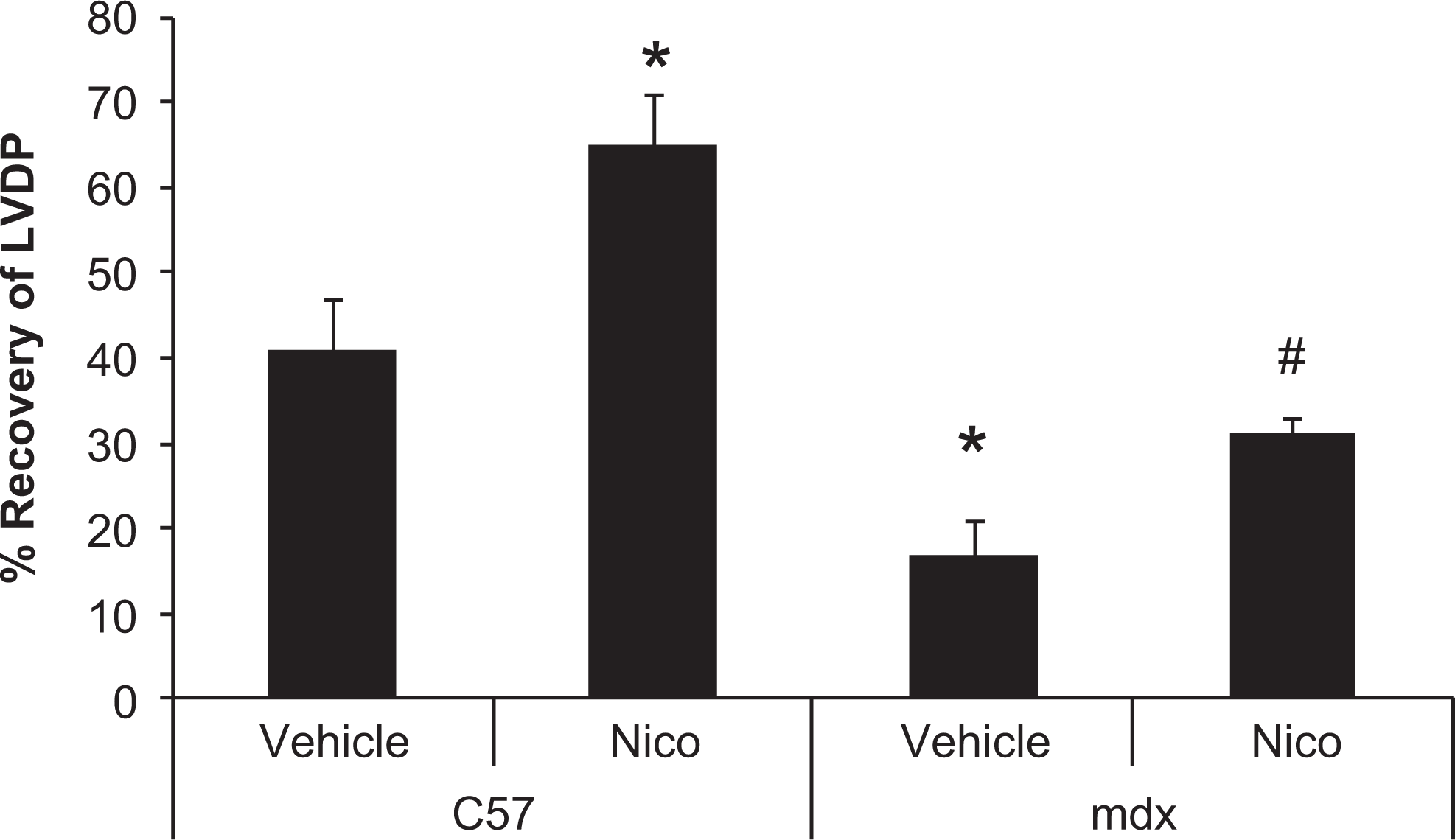
Nicorandil increases recovery of left ventricular developed pressure (LVDP) after ischemia and reperfusion in the isolated mdx heart. Isolated hearts from mdx and C57 control mice were subjected to 30 minutes of global ischemia followed by 2 hours of reperfusion with/without pretreatment with nicorandil (Nico). Nicorandil improves recovery of LVDP in both mdx and control hearts. Data are mean ± standard error of the mean (SEM), n = 8 hearts/group. *P ≤ .05 versus C57 + vehicle, #P ≤ .05 versus mdx-vehicle.
Discussion
This study provides the first evidence that nicorandil has the potential to protect against stress-induced cardiac injury in dystrophin deficiency. We demonstrated that nicorandil ameliorated cell injury and death in dystrophin-deficient human iPSC-derived cardiomyocytes induced by S/R by preventing an increase in ROS production and by maintaining mitochondrial function. In addition, nicorandil improves cardiac function in the isolated mdx mouse heart after global ischemia and reperfusion. Nicorandil has been shown to exert a cardioprotective effect by limiting ROS production and maintaining mitochondrial function in the clinical setting and in animal models. Therefore, it is possible that nicorandil can prevent cardiomyocyte dysfunction and resultant cardiomyopathy in muscular dystrophy.
Nicorandil was initially developed as a vasodilator with potent coronary and peripheral vascular activity and is used as an antianginal agent in patients with severe coronary artery disease or ischemic heart disease. Nicorandil can also benefit the myocardium through its anti-inflammatory, 37,38 antiapoptotic, 39 and antiarrhythmic 40 –42 effects and its ability to protect mitochondrial function. 39 More specifically, it exerts its cardioprotective effects by opening mitochondrial KATP channels, reducing oxidative damage, 4 preserving adenosine triphosphate production 39 and cytochrome c release, 39 modulating neutrophil properties, 8 and attenuating the mitochondrial Ca2+ overload. 43 These beneficial effects of nicorandil are associated with an improvement in cardiac function in patients with heart failure. A recent systematic review and meta-analysis that evaluated the effect of nicorandil in patients with heart failure concluded that nicorandil may be an additional therapeutic agent for heart failure. 44 Although most of these studies included patients with ischemic heart disease, a few studies included 13% to 20% of patients with dilated cardiomyopathy. Treatment was associated with an improvement in diastolic and systolic function and an improvement in New York Heart Association functional class, which corresponds to an improvement in symptoms. Treatment with nicorandil was associated with a statistically significant 65% reduction in all-cause mortality and hospitalization for cardiac causes. 44
Studies have shown that NO donation 45,46 or antioxidant therapy 47,48 has beneficial effects in the mdx mouse models. For example, restoration of the NO-cGMP pathway 3,49,50 with phosphodiesterase-5 inhibitors, such as sildenafil, improved cardiac pathology in mdx mice. 49,50 However, these outcomes have not translated to clinical improvements in patients with Duchenne or Becker muscular dystrophy. 51,52 Explanations for this lack of clinical benefit of restoring the NO-cGMP pathway could be due to (1) an increase in oxidative stress in the heart or (2) beneficial NO actions independent of cGMP signaling that are not restored by phosphodiesterase-5 inhibitors. For instance, oxidative stress can counteract the protective effects of NO through either increased NO scavenging or impairment of the NO-cGMP pathway. 53 –55 As an example from our study, we found that in N-iCMs and Dys2-iCMs nicorandil decreased ROS through an NO-cGMP-dependent manner but in an NO-cGMP-independent manner in Dys1-iCMs. The increased ROS levels observed at baseline in the Dys1-iCMs may negate NO signaling. Furthermore, NO also directly regulates mitochondrial function and bioenergetics and has been linked to the opening of KATP channels. 56 Nicorandil can circumvent these limitations of NO donors or phosphodiesterase-5 inhibitors through its pleiotropic effects not only as an NO donor but also as an antioxidant and a KATP channel opener. KATP channel openers have not previously been studied in muscular dystrophy.
Limitations
Current insights into the pathogenesis of dystrophin-deficient cardiomyopathy are largely based on the mdx mouse model that contains a nonsense mutation in exon 23. Compared with the mdx mouse, we observed some differences in the human Dys-iCMs, which may be species dependent or mutation dependent, or iPSC reprogramming differences. For instance, compared to wild type, ROS levels are increased in mdx hearts 4,50,57 and in the Dys1-iCMs but not in the Dys2-iCMs under control conditions. This may reflect the severity of the dystrophin mutation between the 2 cell lines as Dys1-iCMs have a null mutation that would be analogous to the null mutation in the mdx mouse, whereas the Dys2-iCMs is predicted to express an aberrant internally truncated protein. However, we cannot discount that this may be specific to the reprogrammed iPSC line and not related to the specific mutation. Considering the Dys1 and Dys2 lines are from unrelated patients with separate dystrophin mutations, we conclude that the phenotype observed in the Dys-iCMs is a result of dystrophin mutations. Furthermore, the redox environment of the cell is complex and may differ between individuals depending on the expression levels of several different pro- and antioxidant systems in which we have only examined a small part of the entire system.
In conclusion, we report that dystrophin-deficient iPSC-derived cardiomyocytes are especially susceptible to cellular stress, which leads to rapid loss of the mitochondrial membrane potential, increased ROS production, and increased cell injury and death. These findings provide measurable parameters for evaluating the effect of potential therapeutics on iPSC-derived cardiomyocytes carrying dystrophin gene mutations and show that these consequences can be alleviated by nicorandil. Nicorandil is a drug that exerts a cardioprotective effect by stimulating NO pathways and KATP channels. With the validation of the cardioprotection of nicorandil in the mdx heart, nicorandil represents a potential therapeutic agent for use in the treatment of cardiomyopathy in muscular dystrophy. The safety and efficacy of nicorandil have not yet been established in children, although there are case reports of using nicorandil in children for the treatment of congenital long QT syndrome, an inherited arrhythmia that carries a risk of sudden death. 58 Therefore, before initiating a clinical trial, it would be prudent to further understand the chronic effects of nicorandil on both cardiac and skeletal muscle structure and function in an animal model of Duchenne muscular dystrophy.
Footnotes
Authors’ Note
This work was done at the Medical College of Wisconsin, Milwaukee, WI, USA.
Acknowledgments
The authors would like to thank Zelkjo Bosnjak, PhD, and Scott Canfield, PhD, from the Department of Anesthesiology, Medical College of Wisconsin, for their assistance and advice on pluripotent cell culture, cardiomyocyte differentiation, and assessment of mitochondrial function.
Author Contributions
Muhammad Z. Afzal and Melanie Reiter contributed equally to the work in this manuscript. Muhammad Z. Afzal and Melanie Reiter contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. Courtney Gastonguay contributed to acquisition and critically revised the manuscript. Xuan Guan and Allison D. Ebert contributed to interpretation and critically revised the manuscript. Jered V. McGivern contributed to acquisition and analysis and critically revised the manuscript. David L. Mack and Martin K. Childers contributed to design, contributed to interpretation, and critically revised the manuscript. Jennifer L. Strande contributed to conception and design, contributed to analysis and interpretation, drafted the manuscript, and critically the revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutions of Health K08 Grant Number HL111148, PPG P01GM066730, and by the National Center for Research Resources and the National Center for Advancing Translational Sciences through Grant Number UL1RR031973 to J.L.S. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.