
Abstract
Aim:
Vascular calcification (VC) is thought to be an independent predictor of cardiovascular morbidity and mortality. Intermedin1-53 (IMD) is a cardiovascular protective peptide and can inhibit vascular medial calcification in rats. In this study, we investigated the effect of IMD on atherosclerotic calcification induced by a high-fat diet plus homocysteine (Hcy) and the potential mechanisms.
Methods:
ApoE−/− mice were fed a high-fat diet with Hcy in drinking water to induce atherosclerotic calcification.
Results:
As compared to the high-fat diet alone, Hcy treatment significantly increased atherosclerotic lesion areas and the number of calcified nodules in aortic roots and was reduced by IMD infusion or 4-phenylbutyric acid (PBA) treatment. In vitro, as compared to calcifying medium alone, Hcy treatment further increased alkaline phosphatase activity, calcium content, and calcium nodule number in human aorta vascular smooth muscle cells (HA-VSMCs), all blocked by IMD or PBA pretreatment. Mechanistically, IMD or PBA significantly alleviated endoplasmic reticulum stress (ERS) activation compared with Hcy treatment. In parallel, IMD or PBA attenuated the messenger RNA levels of osteogenic markers and inflammatory cytokines in aortas and their protein levels in lesions of aortic roots. In vitro, Hcy treatment significantly increased the protein levels of osteoblast-like cell markers in primary rat VSMCs and inflammation markers in mouse peritoneal macrophages, all decreased with IMD or PBA pretreatment. Intermedin1-53 pretreatment also markedly reduced the protein levels of ERS markers in rat VSMCs and mouse peritoneal macrophages.
Conclusions:
Intermedin1-53 protects against Hcy-promoted atherosclerotic calcification in ApoE−/− mice by inhibiting ERS.
Introduction
Vascular calcification (VC) is considered an independent predictor of cardiovascular morbidity and mortality 1 and commonly occurs in atherosclerosis, diabetes mellitus, and end-stage renal disease. 2 Intimal calcification is a feature of advanced atherosclerosis. It is highly associated with plaque burden and can predict the risk of plaque instability, myocardial infarction. 3,4 Patients with spotty calcification undergone more severe coronary atherosclerosis and mostly have a history of myocardial infarction, which confirms that VC contributes to the vulnerability of plaque rupture. 5 Many osteogenic regulatory factors such as osteopontin (OPN), osteocalcin (OCN), bone morphogenetic protein (BMP), and matrix Gla protein are expressed in atherosclerotic lesions. 6 -8 Although initially considered a passive precipitation of crystals, atherosclerotic calcification was found as an active cell-driven process similar to bone development. 9 Genetic fate mapping studies in mouse models have identified that vascular smooth muscle cells (VSMCs) are a major cell source of osteochondrogenic differentiation and contributed to atherosclerotic intimal calcification. 10,11 However, the underlying mechanisms that regulate VSMC transdifferentiation are not well understood.
The endoplasmic reticulum (ER) is an organelle containing-membrane structure responsible for protein folding, lipid biosynthesis, and calcium homeostasis. Various cellular stressors can lead to accumulation of unfolded and misfolded proteins in ER, which triggers a response termed as endoplasmic reticulum stress (ERS). ERS initially is an adaptive response but prolonged or overwhelming ERS can induce apoptotic cell death. 12 Accumulating evidences have demonstrated that ERS is implicated in the development and progression of cardiovascular diseases, particularly atherosclerosis and VC. 12,13 Endoplasmic reticulum stress contributes to intimal calcification by promoting osteogenic differentiation of VSMCs in the ApoE−/− mouse model induced by chronic kidney disease. 14,15 In addition, ERS can regulate inflammation by activating the pathways c-Jun N-terminal kinase (JNK) and nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB). 16 Moreover, inflammation is a key contributor to atherosclerotic calcification. 17 -19 Therefore, inhibiting ERS may be a promising strategy to treat atherosclerotic calcification.
Homocysteine (Hcy) is a sulfur-containing amino acid from methionine, and hyperhomocysteinemia (HHcy) is an independent risk factor for atherosclerosis. 20 Hyperhomocysteinemia as a potent pro-inflammatory factor can accelerate atherosclerosis in ApoE−/− mice by activating an inflammatory response 21 and ERS. 22 In patients with hyperlipidemia, mild HHcy is associated with aortic calcification. 23 In patients undergoing percutaneous coronary interventions, the risks of long-term major adverse cardiac events were markedly higher when Hcy level was elevated. 24 These researches suggest that Hcy may accelerate hyperlipidemia-induced atherosclerotic vascular disease. Furthermore, Hcy could induce ERS and potentiate calcification of cultured rat aortic SMCs. 25,26
Recently, numerous studies revealed that vasoactive peptides such as adrenomedullin (ADM), C-type natriuretic peptide, and parathyroid hormone-related peptide are involved in vascular calcification. 27 Intermedin1-53 (IMD), also known as ADM2, which belongs to the calcitonin/calcitonin gene-related peptide family, is a potential endogenous protective peptide of the cardiovascular system. 28 We and others found that IMD administration can inhibit atherosclerosis in ApoE−/− mice 29 -31 and vascular medial calcification in rats. 32,33 We also reported that the level of IMD in calcified aortas of rats was downregulated 32,33 and IMD treatment could alleviate VSMC calcification by inhibiting ERS. 34 Moreover, we found that IMD could protect against myocardial fibrosis induced by high-fat diet plus Hcy in ApoE−/− mice. 35 However, whether IMD attenuates atherosclerotic calcification induced by high-fat diet plus Hcy in ApoE−/− mice is still unknown. In this study, we investigated the effect of IMD on a mouse model of atherosclerotic calcification induced by a high-fat diet plus Hcy and the possible mechanisms in ApoE−/− mice.
Methods and Materials
Materials
Synthetic human IMD1-53 was from Phoenix Pharmaceuticals (Burlingame, California). Alzet Mini-osmotic Pumps (model 2006) were from DURECT Corp (Cupertino, California). DL-homocysteine (DL-Hcy), 4-phenylbutyric acid (PBA), and Oil-red O were from Sigma-Aldrich (St Louis, Missouri). Primary antibodies for glucose-regulated protein78 (GRP78, ab21865), activating transcription factor (ATF4, ab216839), cleaved-ATF6 (ab65838), p-inositol-requiring kinase 1 alpha (p-IRE1α, ab48187), CCAAT/enhancer-binding protein homologous protein (CHOP, ab10444), alkaline phosphatase (ALP, ab95462), smooth muscle 22α (SM22α, ab10135), α-smooth muscle actin (α-actin, ab21027), Runt-related transcription factor 2 (Runx2, ab23981), bone morphogenetic protein 2 (BMP2, ab14933), CD68 (ab955), NF-κB p65 (ab90532), monocyte chemoattractant protein 1 (MCP-1, ab25124), and tumor necrosis factor α (TNFα, ab6671) were from Abcam PLC (Cambridge, United Kingdom). Primary antibodies for osteopontin (OPN, sc-10593), β-actin (sc-47778), GAPDH (sc-25778), and all secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, California). The 1% Alizard Red S was from Leagene Biotech Co (Beijing, China). The kit for reverse transcription of RNA and SuperReal PreMix Plus for real-time polymerase chain reaction (PCR) were from TIANGEN Biotech (Beijing, China).
Animals
Eight-week-old male ApoE−/− mice were provided by the Animal Center of Peking University Health Science Center (Beijing, China). All animals were fed a high-fat diet (21% lard and 0.15% cholesterol) for 16 weeks. After 10 weeks, all ApoE−/− mice were randomly divided into 4 groups: (1) high-fat diet group (HF, n = 10): normal drinking water; (2) HF plus Hcy group (HF + Hcy, n = 10): 1.8 g/L DL-Hcy was added into drinking water for 6 weeks; (3) HF plus Hcy plus IMD group (HF + Hcy + IMD, n = 10): 300 ng/kg/h IMD1-53 was dissolved in sterile saline and subcutaneously administered via Alzert mini-osmotic pumps (model 2006, Cupertino, California) for 6 weeks at the same time as the Hcy treatment described above; or (4) HF plus Hcy plus 4-phenylbutyric acid group (HF + Hcy + PBA, n = 10): PBA (100 mg/kg dissolved in sterile saline) intragastrically administered for 6 weeks at the same time as the Hcy treatment described above. At the end of the experiment, all animals were anesthetized with 1% pentobarbital sodium and killed by removing the eyeball and taking blood. The animal care and experimental protocols were conducted by following the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011) and were approved by the Animal Care Committee of Peking University Health Science Center.
Cell Culture
Peritoneal macrophages were isolated as described 29 with minor modification; 8-week-old male C57BL/6J mice were injected intraperitoneally with 1 mL 3% brewer thioglycollate medium (BD, cat.no.211716). Four days later, mice were killed by cervical dislocation; macrophages were obtained by peritoneal lavage with 10 mL serum-free cold Dulbecco’s modified Eagle medium (DMEM; Gibco, 12800-017, Waltham, Massachusetts). Cells were plated at 1.0 × 106 per mL DMEM with 10% fetal bovine serum (FBS) for 2 hours at 37°C, then nonadherent cells were washed off, and adherent cells were prepared for experiments.
The explant culture of rat primary VSMCs was performed as described previously. 36 Briefly, male Sprague-Dawley rats (120-150 g) were killed by cervical dislocation. Thoracic aortas were isolated, removed endothelium and adventitia, and cut into small pieces, then placed in DMEM containing 20% FBS. Using α-actin staining of cultured cells to confirm a positive response. Vascular smooth muscle cells at passages 5 to 10 were used for experiments.
The HA-VSMCs (ATCC, CRL-1999) or rat primary VSMCs were cultured in DMEM with 10% FBS under 95% air and 5% CO2 at 37°C. To induce calcification, HA-VSMCs or rat primary VSMCs were incubated in calcifying medium containing 2.5 mmol/L Ca+ (0.7 mmol/L CaCl2 was added into DMEM containing 1.8 mmol/L CaCl2) and 5 mmol/L β-glycerophosphate disodium salt hydrate with 10% FBS.
Ultrasonography
The day before the end of the experiment, mice were anesthetized with 1% pentobarbital sodium (60 mg/kg) by intraperitoneal injection and underwent ultrasonography by use of the Vevo 770TM Imaging System (Visual Sonics, Toronto, Canada) at 30 MHz to visualize the abdominal aorta. The luminar diameter and intima-media thickness of the abdomimal aorta posterior to the liver were measured.
Lipid Assay
Blood samples were taken by removing the eyeball of mice; plasma was obtained by centrifugation at 3000 rpm for 15 minutes at 4°C. Plasma levels of total cholesterol (TC), triglycerides (TG), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) were measured by using kits from Zhong Sheng Bio-technology (Beijing, China).
Western Blot Analysis
Aorta tissue or cell lysates were prepared by using lysis buffer. Equal amounts of protein samples were loaded for sodium dodecyl sulphate–polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. After incubation in 5% nonfat milk for 1 hour, membranes were incubated with primary antibodies for GRP78 (1:2000), ATF4 (1:500), cleaved-ATF6 (1:300), p-IREα (1:500), CHOP (1:500), SM22α (1:1000), α-actin (1:2000), Runx2 (1:1000), BMP2 (1:500), NF-κB (1:1000), MCP-1 (1:500), TNFα (1:1000), GAPDH (1:1000), or β-actin (1:4000) overnight at 4°C, and with horseradish peroxidase–conjugated secondary antibody for 1 hour at room temperature. Protein bands were visualized by ECL. Protein levels were analyzed by using NIH image software and normalized to that of β-actin or GAPDH. Except the β-actin, GAPDH, and all secondary antibodies from Santa Cruz Biotechnology, all other primary antibodies were from Abcam PLC.
Quantitative Real-Time Polymerase Chain Reaction Analysis
Total RNA was extracted from aortic tissue by using Trizol reagent. RNA of 2.0 µg was reverse transcribed to complementary DNA (cDNA) by using M-MLV and an oligo (dT) primer. The 7500 Fast Real-Time PCR System (Applied Biosystems, Waltham, Massachusets) was used to amplify cDNA. The amount of PCR product in each cycle was evaluated by Eva Green fluorescence. The 2−ΔΔCt method was used to analyze relative messenger RNA (mRNA) levels, with GAPDH as a reference. The primers for real-time PCR are in Supplemental Table 1.
Histology
Mice were killed and arterial roots with the heart were removed, fixed in 4% paraformaldehyde overnight, and embedded in paraffin or optimal cutting temperature compond for frozen sections, and 7-µm cross sections were prepared. Paraffin sections were used for hematoxylin and eosin (H&E) and von Kossa staining and immunostaining, and frozen sections were used for Oil-red O and immunofluorescence staining.
Von Kossa Staining and Alizarin Red Staining
Von Kossa staining was performed to observe atherosclerotic calcification in aortic roots. Briefly, aortic paraffin sections were dewaxed and dehydrated, then incubated in 1% silver nitrate solution for 2 hours under intense sunlight, washed, and immersed in sodium thiosulfate for 2 minutes and counterstained with aldehyde fuchsin. The brown/black color represents calcified nodules.
For Alizarin red S staining, aortic paraffin sections were dewaxed and dehydrated, then incubated in 1% Alizarin red S solution (pH 4.2) for 10 minutes, washed 3 times with deionized water; HA-VSMCs were cultured in 6-well plates for 10 days with media changes every 2 days. After removing medium, cells were washed 3 times with cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 15 minutes, then washed 3 times with deionized water, and exposed to 1% Alizarin-red S solution (pH 4.2) for 30 minutes, washed 3 times again with deionized water, and one time quickly with 0.2% acetic acid; calcified lesions or calcified cells were stained a red/purple color.
Quantification of Calcium Content and Alkaline Phosphatase Activity Assay
The HA-VSMCs were cultured in normal medium or calcifying medium for 8 days with media changes every 2 days. After removing the medium, cells were washed 3 times with cold PBS and treated with 0.6 mol/L HCl overnight at 4°C. Cell lysates were centrifuged and calcium content in the supernatant was measured by colorimetry with a calcium kit (Biosino Bio-Technology and Science, Beijing, China). The data were normalized to the total protein concentration.
For ALP activity assay, HA-VSMCs were cultured in normal medium or calcifying medium for 3 days. Cell lysates were prepared and ALP activity was determined by using an ALP assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Immunohistochemistry and Immunofluorescence Staining
For immunohistochemistry staining, paraffin sections (7 µm) from mice arterial roots were blocked with 10% normal goat or rabbit serum for 1 hour at 37°C and incubated at 4°C overnight with primary antibodies for ATF4 (1:200), cleaved-ATF6 (1:200), p-IRE1α(1:50), CHOP (1:50), BMP2 (1:100), Runx2 (1:100), ALP (1:50), or MCP-1 (1:100), then incubated with secondary antibodies for 1 hour and stained with Diaminobenzidine. All primary antibodies were from Abcam PLC, and all secondary antibodies were from Santa Cruz Biotechnology.
For immunofluorescence staining, frozen sections (7 µm) from mouse arterial roots were blocked with 10% normal goat or rabbit serum for 1 hour at 37°C and incubated at 4°C overnight with primary antibodies for CD68 (Abcam PLC, 1:200) or OPN (Santa Cruz Biotechnology, 1:50), then with DyLight-labeled secondary antibodies (EarthOx, LLC, San Francisco, California) for 1 hour. Nuclei were counterstained with Hoechst 33342 for 5 minutes. Fluorescence signals were observed under a fluorescence-inverted microscope (Leica Imaging Systems, Cambridge, United Kingdom).
Statistical Analysis
All data are expressed as mean ± standard deviation. GraphPad Prism v5.01 was used to analyze data. Student t test was used to compare 2 groups and 1-way analysis of variance, followed by Newman–Keuls test for more than 2 groups. P < .05 was considered statistically significant.
Results
Intermedin1-53 Ameliorated Hcy-Accelerated Atherosclerosis
ApoE–/– mice were fed a high-fat diet (16 weeks) with Hcy in drinking water for 6 weeks to induce atherosclerosis and atherosclerotic calcification. First, we determined changes in endogenous mRNA levels of IMD and its receptors CRLR, RAMP1/2/3 in aortas. In Hcy plus HF-treated mice versus HF alone, the mRNA level of IMD was reduced by 66% (P < .05; Supplemental Figure 1A) and those of CRLR and RAMP3 were significantly increased by 1.66- and 2.7-fold (all P < .05), respectively (Supplemental Figure 1B and C). The mRNA expression of RAMP1 and RAMP2 was higher but not significant in Hcy plus HF-than HF-treated mice (Supplementary Figure 1C).
Intermedin1-53 can reduce atherosclerosis in ApoE–/– mice under a normal chow diet 29,31 or a high-fat diet, 30,37 so we investigated whether IMD has a protective role in atherosclerosis induced by a high-fat diet plus Hcy. After 16 weeks of a high-fat diet, the body weight did not differ among mouse groups (Figure 1A). Plasma TG, TC, and LDL-C levels were higher with Hcy than HF alone, and IMD treatment significantly decreased the Hcy-increased TG, TC, and LDL-C levels (Table 1). On ultrasonography, Hcy treatment decreased the luminar diameter, although not significantly, and significantly increased intima-media thickness of abdominal aortas as compared to HF treatment alone; these changes were rescued by IMD treatment (Figure 1B-D). On Oil-red O and H&E staining, the plaque area in aortas and aortic roots was greater with Hcy treatment than HF alone, and the increased plaque area was attenuated by IMD treatment (Figure 1E and F).
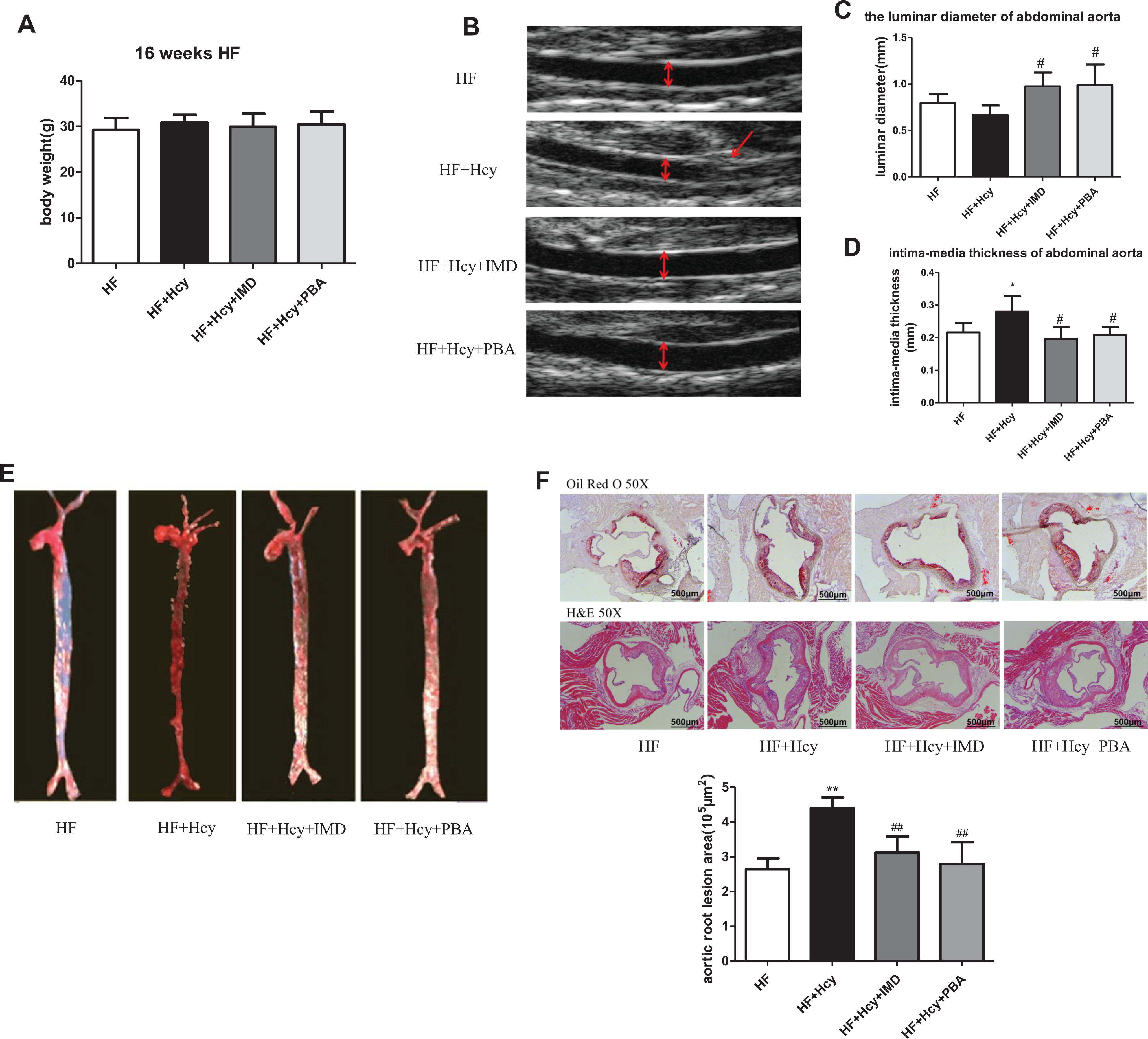
IMD1-53 ameliorated Hcy-accelerated atherosclerosis. Eight-week-old ApoE–/– mice were fed a high-fat diet for 16 weeks with different treatments. A, The body weight of mice after the high-fat diet for 16 weeks (n = 10 per group). B to D, Luminar diameter and intima-media thickness of abdominal aorta by ultrasonography (n = 5 per group). E, Representative lesion areas of enface aortas stained by Oil-red O. F, Representative Oil-red O and H&E staining of aortic root sections and quantification of aortic root lesion areas (n = 5-7 per group). Scale bar, 500 μm. Data are mean ± SD. *P < .05, **P < .01 versus HF. #P < .05, ##P < .01 versus HF + Hcy. H&E indicates hematoxylin and eosin; Hcy, homocysteine; HF, high fat; IMD1- 53, intermedin1- 53; SD, standard deviation.
Effect of Intermedin (IMD) on Plasma Lipid Levels of ApoE–/– mice.a

Abbreviations: Hcy, homocysteine; HDL, high-density lipoprotein cholesterol; HF, high fat; LDL-C, low-density lipoprotein cholesterol; PBA, 4-phenylbutyric acid; SD, standard deviation; TC, total cholesterol; TG, triglyceride.
a Data are shown as mean ± SD.
b P < 05 versus HF.
c P < .05 versus HF + Hcy.
d P < .01 versus HF.
e P < .01 versus HF + Hcy.
Intermedin1-53 Ameliorated Hcy-Accelerated Atherosclerotic Calcification and VSMC Calcification
We have previously shown that IMD can inhibit vascular medial calcification in rats, 32,33 but whether IMD has a protective role in atherosclerotic calcification is unknown. On immunostaining, Hcy markedly increased the expression of ALP, an important marker of vascular calcification, in lesions of aortic roots, which could be inhibited by IMD (Figure 2A). Alizarin red S staining and von Kossa staining showed that calcium-phosphate salt deposition in lesions of aortic roots, an indication of atherosclerotic calcification, was accelerated by Hcy and attenuated by administration of IMD (Figure 2B-E).
To further confirm the results in vivo, we observed the effect of IMD on cultured HA-VSMCs. Alkaline phosphatase activity and calcium content were significantly higher with Hcy treatment than calcifying medium alone and could be blocked by IMD pretreatment (Figure 2F and G). Moreover, calcium-phosphate salt deposition was accelerated by Hcy treatment but significantly decreased with IMD pretreatment (Figure 2H and I). These results suggest that the administration of IMD could ameliorate Hcy-accelerated atherosclerotic calcification and VSMC calcification.
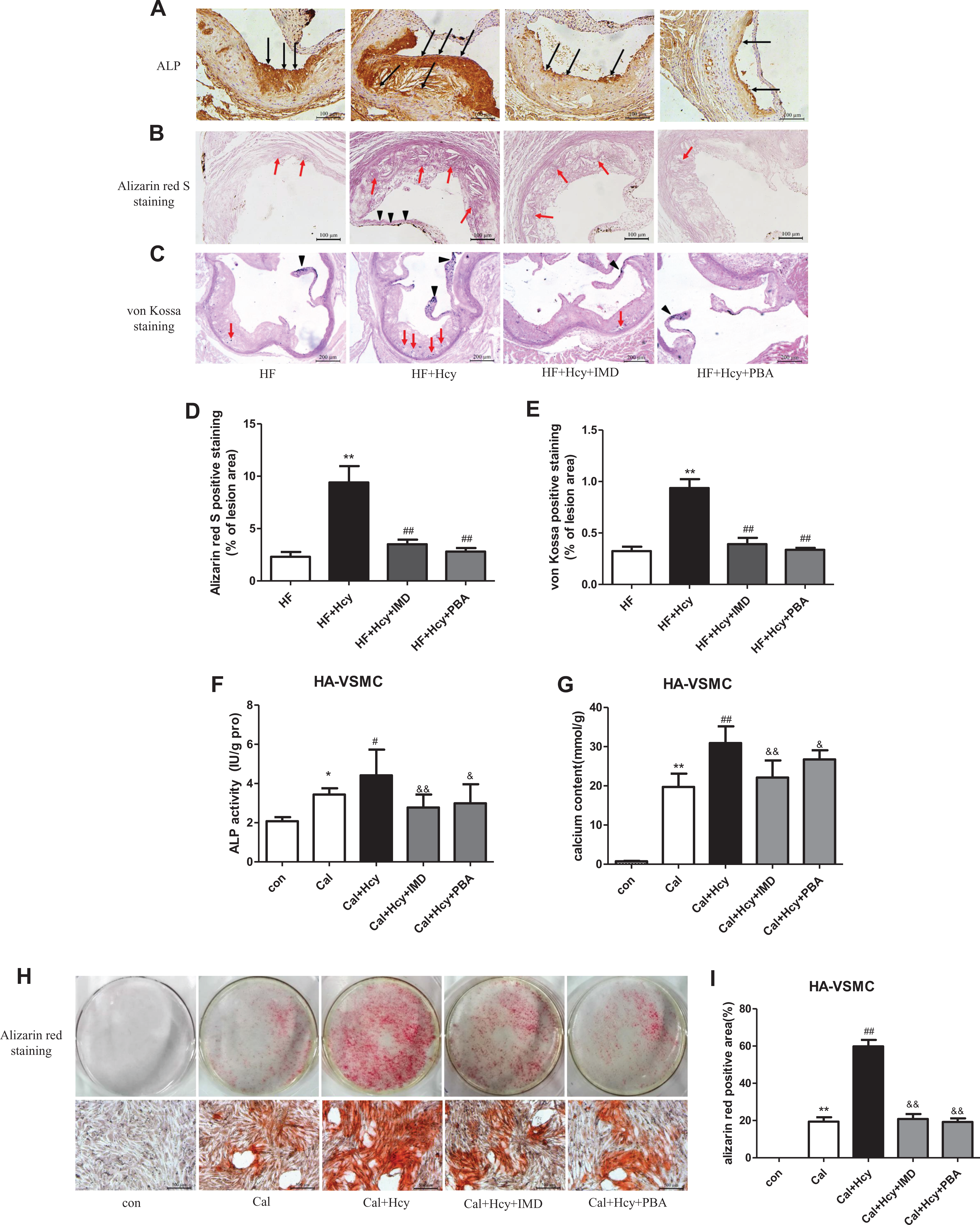
IMD1-53 ameliorated Hcy-accelerated atherosclerotic calcification in vivo and VSMC calcification in vitro. A to E, Eight-week-old ApoE–/– mice were fed a high-fat diet for 16 weeks. A, Representative immunohistochemical staining of alkaline phosphatase (ALP) in aortic root sections of ApoE–/– mice. Scale bar, 100 μm. B, Representative Alizarin red S staining in aortic root sections of ApoE–/– mice, the red/purple color represents calcium deposits. The red line arrow indicates calcium deposits in lesions, and black arrow indicates calcium deposits in valve. Scale bar, 100 μm. C, Representative von Kossa staining in aortic root sections of ApoE–/– mice; the red line arrow indicates calcified nodules in lesions, and black arrow indicates calcified nodules in valve. Scale bar, 200 μm. D and E, The quantification of Alizarin red S staining and von Kossa staining in aortic root sections of ApoE–/– mice. Data are mean ± SD, n = 4. **P < .01 versus HF. ##P < .01 versus HF + Hcy. F to I, HA-VSMCs were cultured in normal media (con) or calcifying media only (Cal) or calcifying media treated with 100 μmol/L Hcy (Cal + Hcy), or calcifying media treated with 100 μmol/LHcy plus 10−7 mol/L IMD (Cal + Hcy + IMD) or calcifying media treated with 100 μmol/L Hcy plus 5 mmol/L PBA (Cal + Hcy + PBA)for 3 days for ALP activity assay (F), for 8 days for calcium content assay (G) or for 10 days for Alizarin red S staining (H and I). Scale bar, 500 μm. Data are mean ± SD, n = 6. *P < .05, **P < .01 versus con. #P < .05, ##P < .01 versus Cal. &P < .05, &&P < .01 versus Cal + Hcy. HF indicates high fat; IMD1- 53, intermedin1- 53; HA-VSMC, human aorta vascular smooth muscle cell; Hcy, homocysteine; PBA, 4-phenylbutyric acid; SD, standard deviation.
Intermedin1-53 Ameliorated Atherosclerotic Calcification by Reversing Hcy-Promoted ERS
Endoplasmic reticulum stress is involved in vascular calcification 13 -15 and our previous study reported that IMD can inhibit ERS in calcified VSMCs in vitro. 34 Moreover, Hcy could exacerbate atherosclerosis by inducing ERS, and ERS inhibitors could suppress atherosclerosis induced by Hcy. 38 Therefore, we investigated whether IMD could mitigate Hcy-induced ERS. The protein expression of the ERS markers ATF4, cleaved-ATF6, p-IRE1α, and CHOP was significantly increased in atherosclerotic lesions of ApoE–/– mice with Hcy treatment, as indicated by immunohistochemistry staining, and IMD reduced these Hcy-promoted protein levels (Figure 3A-D). Thus, IMD could reverse Hcy-promoted ERS.

IMD1-53 ameliorated atherosclerotic calcification by reversing Hcy-promoted ERS. Eight-week-old ApoE–/– mice were fed a high-fat diet for 16 weeks with different treatments. A to D, Representative immunohistochemical staining of ATF4, cleaved-ATF6, p-IRE1α, and CHOP in aortic root sections of ApoE–/– mice. Scale bar, 100 μm. ATF indicates activating transcription factor; CHOP, CCAAT/enhancer-binding protein homologous protein; Hcy, homocysteine; IMD1- 53, intermedin1- 53.
To further ascertain whether IMD-attenuated atherosclerotic calcification by inhibiting ERS, we tested the role of PBA, a chemical chaperone, which is known to inhibit ERS, 39 in mice. As expected, PBA markedly inhibited the Hcy upregulated the protein expression of ERS markers in plaques (Figure 3A-D). Meanwhile, ALP expression and calcium-phosphate salt deposition were significantly reduced in lesions with PBA treatment as compared to HF plus Hcy treatment (Figure 2A-E). In vitro, ALP activity and calcium content in HA-VSMCs was decreased by PBA pretreatment as compared to Hcy treatment (Figure 2F and G). Finally, PBA pretreatment could reduce calcium-phosphate salt deposition accelerated by Hcy in HA-VSMCs (Figure 2H and I). These results suggested that atherosclerotic calcification could be ameliorated by inhibiting ERS and that IMD could alleviate Hcy-accelerated atherosclerotic calcification at least in part by inhibiting ERS.
Intermedin1-53 Inhibited Osteoblastic Differentiation of VSMCs by Attenuating ERS In Vivo and In Vitro
The osteochondrogenic transdifferentiation of VSMCs is a vital characteristic in the development of vascular calcification and involves loss of VSMC lineage markers and gain of osteoblast markers. 40 Genetic fate-mapping studies in mouse models have also shown that VSMCs are a major cell source of osteochondrogenic differentiation and contributed to atherosclerotic intimal calcification. 10,11 The mRNA expression of osteogenic markers BMP2, cbfα1, osterix, and OCN in aortas was significantly upregulated by Hcy treatment as compared to HF treatment alone and was reversed by IMD administration (Figure 4A). Meanwhile, the protein levels of osteogenic markers BMP2, Runx2, and OPN were markedly increased in lesions of HF plus Hcy treatment mice and were reduced by IMD treatment (Figure 4B-D).
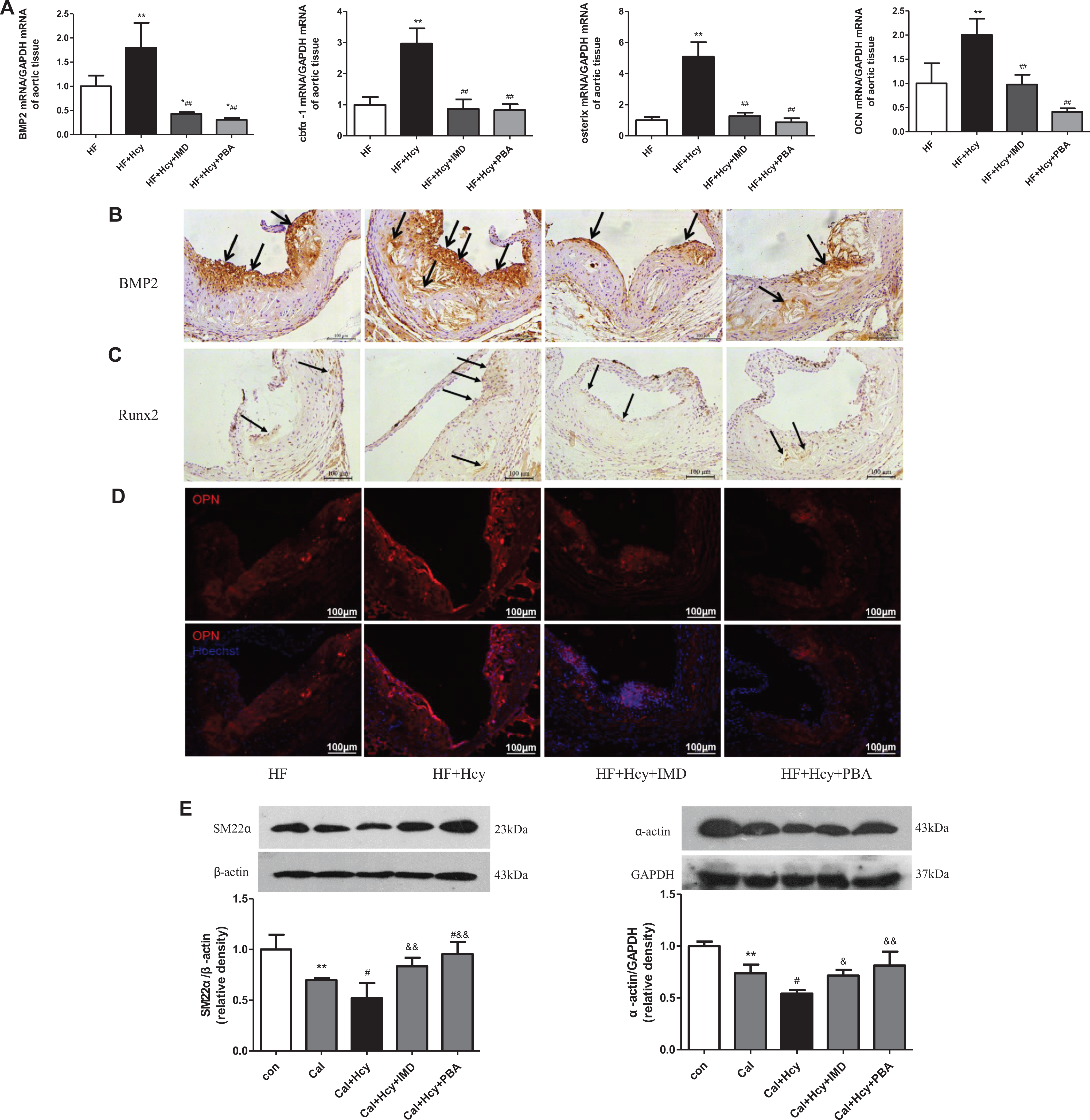
IMD1-53 inhibited osteoblastic differentiation of VSMCs by attenuating ERS in vivo and in vitro. A to D, Eight-week-old ApoE–/– mice were fed a high-fat diet for 16 weeks. A, Quantitative real-time PCR analysis of mRNA levels of bone morphogenetic protein 2 (BMP2), core-binding factor α1 (cbfα-1), osterix, and osteocalcin (OCN). Data are mean ± SD, n = 3. *P < .05, **P < .01 versus HF. ##P < .01 versus HF + Hcy. B and C, Representative immunohistochemical staining of BMP2 and Runt-related transcription factor 2 (Runx2) in aortic root sections of ApoE–/– mice. Scale bar, 100 μm. D, Representative immunofluorescence staining of osteopontin (OPN) in aortic root sections of ApoE–/– mice. Scale bar, 100 μm. E to G, primary rat VSMCs were cultured in normal media (con) or calcifying media only (Cal) or calcifying media treated with 100 µmol/L Hcy (Cal + Hcy), or calcifying media treated with 100 µmol/LHcy plus 10−7 mol/L IMD (Cal + Hcy + IMD), or calcifying media treated with 100 µmol/L Hcy plus 5 mmol/L PBA (Cal + Hcy + PBA) for 6 days. Western blot analysis of the protein levels of SM22α and α-actin (E), BMP2 and Runx2 (F), and GRP78, cleaved-ATF6, p-IRE1α, and CHOP (G). Data are mean ± SD, n = 3 to 4. *P < .05, **P < .01 versus con. #P < .05, ##P < .01 versus Cal. &P < .05, &&P < .01 versus Cal + Hcy. ATF indicates activating transcription factor; CHOP, CCAAT/enhancer-binding protein homologous protein; Hcy, homocysteine; HF, high fat; IMD1- 53, intermedin1- 53; PBA, 4-phenylbutyric acid; SD, standard deviation; SM22α, smooth muscle 22α; VSMC, vascular smooth muscle cell.

To confirm that IMD could inhibit osteoblast differentiation of VSMCs potentiated by Hcy and investigate the underlying mechanisms, we studied the role of IMD in primary rat VSMCs. As compared to calcifying rat VSMCs, Hcy treatment further decreased the protein levels of the VSMC lineage markers SM22α and α-actin, which were reversed by IMD pretreatment (Figure 4E). Moreover, IMD pretreatment markedly reduced the Hcy-increased protein levels of the osteogenic markers BMP2 and Runx2 (Figure 4F) and ERS markers GRP78, cleaved ATF6, p-IRE1α, and CHOP in rat VSMCs (Figure 4G), which agrees with the change in osteoblastic markers.
To investigate whether IMD inhibits osteoblastic differentiation of VSMCs by attenuating ERS, we determined the effect of PBA on osteoblastic differentiation in vivo and in vitro. In vivo, administration of PBA inhibited Hcy-promoted mRNA expression of osteogenic markers BMP2, cbfα1, osterix, and OCN in aortas and Hcy-induced protein levels of BMP2, Runx2, and OPN in lesions (Figure 4A-D). In vitro, PBA pretreatment reversed the Hcy-decreased protein levels of SM22α and α-actin and Hcy-increased protein levels of Runx2 and BMP2 in rat VSMCs (Figure 4E-F). These data suggested that IMD inhibited osteoblastic differentiation of VSMCs at least in part by attenuating ERS.
Intermedin1-53 Alleviated Inflammation of Macrophages by Attenuating ERS In Vivo and In Vitro
Studies revealed that inflammation could promote osteogenesis 18 in atherosclerotic plaques and atherosclerotic calcification. 17 In this study, we found that Hcy upregulated the mRNA expression of inflammatory marker such as IFN-γ, IL-6, MCP-1, TNFα in aortas, and administration of IMD inhibited the Hcy-induced upregulation (Figure 5A). Immunostaining revealed markedly increased infiltration of CD68-positive macrophages in lesions of aortic roots after Hcy treatment, which was reduced by IMD infusion (Figure 5B). In addition, MCP-1 was highly expressed in plaques with Hcy treatment, and its level was decreased by IMD infusion (Figure 5C).
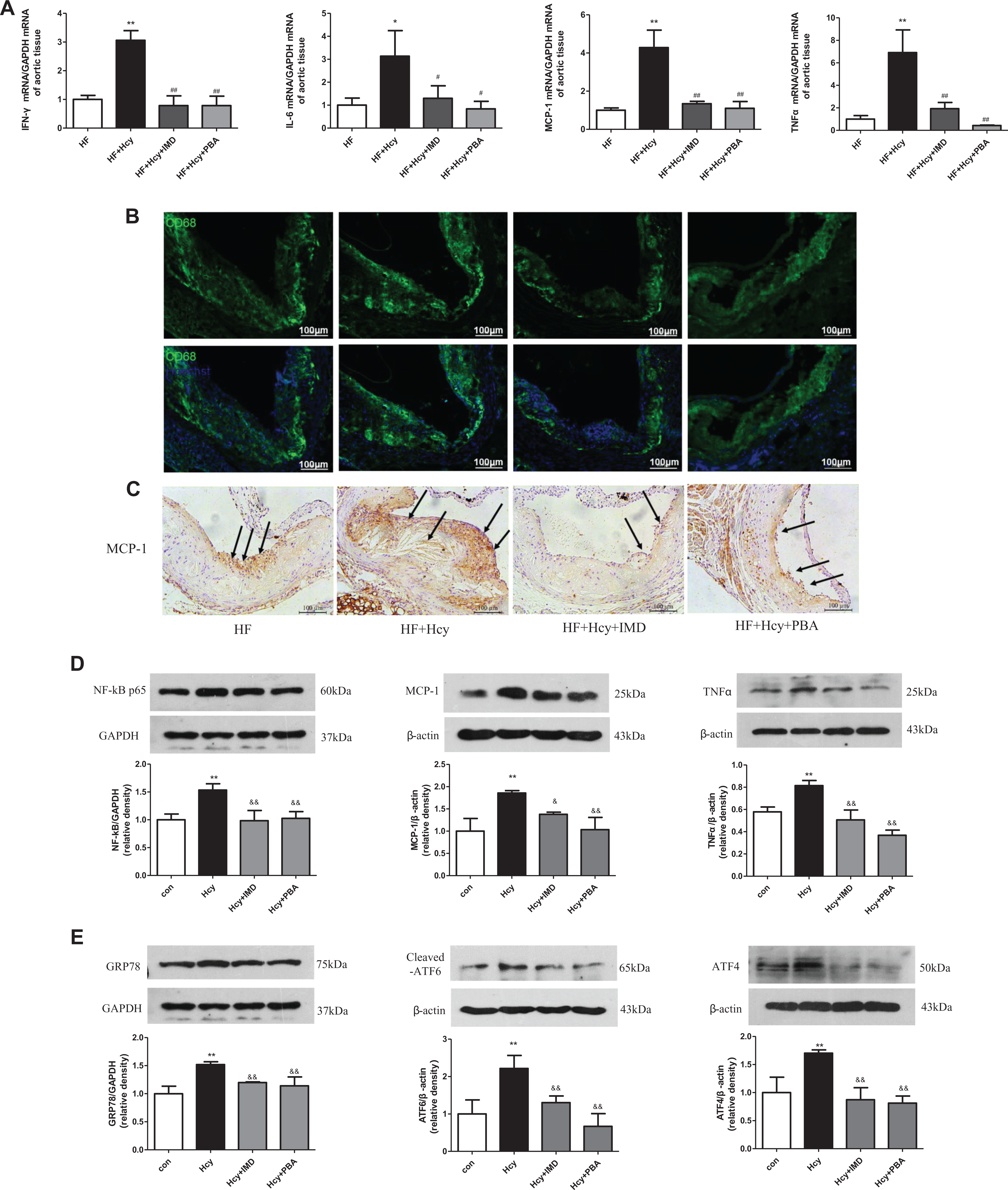
IMD1-53 alleviated inflammation of macrophages by attenuating ERS in vivo and in vitro. A to C, Eight-week-old ApoE–/– mice were fed a high-fat diet for 16 weeks. A, Quantitative real-time PCR analysis of mRNA levels of interferon-γ (IFNγ), interleukin-6 (IL-6), monocyte chemotactic protein-1 (MCP-1), and tumor necrosis factor α (TNFα). Data are mean ± SD, n = 3. *P < .05, **P < .01 versus HF. #P < .05, ##P < .01 versus HF + Hcy. B, Representative immunofluorescence staining for CD68 in aortic root sections of ApoE–/– mice. Scare bar, 100 μm. C, Representative immunohistochemical staining of MCP-1 in aortic root sections of ApoE–/– mice. Scale bar, 100 μm. D and E, Peritoneal macrophages from C57BL/6J were cultured in DMEM without treatment (con) or with 300 µmol/L Hcy (Hcy), or with 300 µmol/L Hcy plus pretreatment for 1 hour with 10−7 mol/L IMD (Hcy + IMD), or with 300 µmol/L Hcy plus pretreatment for 1 hour with 5 mmol/L PBA (Hcy + PBA) for 24 hour. Western blot analysis of the protein levels of NF-κB p65, MCP-1 and TNFα (D), and GRP78, cleaved-ATF6 and ATF4 (E). Data are mean ± SD, n = 3. **P < .01 versus con. &P < .05, &&P < .01 versus Hcy. ATF indicates activating transcription factor; Hcy, homocysteine; HF, high fat; IMD1- 53, intermedin1- 53; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PBA, 4-phenylbutyric acid; SD, standard deviation.
To confirm that IMD could inhibit inflammation induced by Hcy and the potential mechanisms, we investigated the effect of IMD on primary peritoneal macrophages isolated from C57BL/6J mice. The protein levels of NF-κB p65, MCP-1, and TNFα were significantly increased in peritoneal macrophages with Hcy treatment and were blocked by IMD pretreatment (Figure 5D). Meanwhile, IMD pretreatment markedly reduced the Hcy-upregulated protein levels of ERS markers GRP78, cleaved ATF6, and ATF4 in peritoneal macrophages (Figure 5E), which agrees with the change in inflammation markers. Therefore, IMD may inhibit Hcy-induced inflammation by attenuating ERS.
Endoplasmic reticulum stress has been proposed to mediate the inflammatory response by inducing reactive oxygen species production, activating inflammatory pathways such as NF-κB, JNK, and IRE3. 16 To confirm our hypothesis that IMD could inhibit Hcy-induced inflammation by attenuating ERS, we investigated the effect of PBA on inflammation in vivo and in vitro. 4-Phenylbutyric acid administration inhibited the Hcy-induced mRNA expression of IFN-γ, IL-6, MCP-1, and TNFα in aortas and the Hcy-induced protein levels of NF-κB p65, MCP-1, and TNFα in primary peritoneal macrophages (Figure 5A and D). Moreover, macrophage infiltration and MCP-1 expression in lesions were significantly reduced with PBA than Hcy treatment (Figure 5B and C). These results indicate that alleviating ERS could inhibit the Hcy-induced inflammation, and IMD inhibited the inflammation of macrophages at least in part by attenuating ERS.
Discussion
Our study uncovered the critical roles of IMD in atherosclerotic calcification. ApoE–/– mice with a high-fat diet plus Hcy treatment showed increased lesion sizes and formation of calcified nodules in aortic roots, along with activated ERS, promoted osteoblastic differentiation, and increased inflammation. Moreover, endogenous IMD mRNA level was reduced in aortas with Hcy treatment, but the mRNA levels of CRLR/RAMP3 receptor complex were increased. Administration of exogenous IMD reduced Hcy-promoted lesion size and atherosclerotic calcification by inhibiting ERS. Furthermore, IMD treatment inhibited Hcy-accelerated osteoblastic differentiation of VSMCs, infiltration of macrophages, and expression of MCP-1 in aortic root lesions, which might be mediated by inhibiting ERS.
Clinical and epidemiological studies have demonstrated that elevated plasma Hcy level is a potent risk factor for arteriosclerosis 20 and atherosclerotic complications of end-stage renal disease. 41 In patients with hyperlipidemia, mild HHcy was associated with aortic calcification, 23 and administration of exogenous Hcy accelerated atherosclerosis in ApoE–/– mice, 42 which suggest that Hcy may accelerate hyperlipidemia-induced atherosclerotic vascular disease. In this study, we used ApoE–/– mice fed a high-fat diet for 16 weeks, with Hcy treatment for 6 weeks after 10 weeks of the diet to establish the atherosclerotic calcification model. Atherosclerotic lesion size was markedly increased by Hcy treatment, which agrees with previous studies. 42 Meanwhile, administration of Hcy increased the formation of calcified nodules in intimal plaque of aortic roots, an indication of atherosclerotic calcification. Then, we examined the changes in mRNA level of IMD and its receptors. Consistent with the results of IMD in vascular calcification in rats induced by 5/6 nephrectomy plus vitamin D3, 32 IMD mRNA level was significantly reduced and levels of the CRLR/RAMP3 receptor complex were significantly increased in Hcy-treated mouse aortas. However, in the rat vascular calcification model induced by vitamin D3 plus nicotine, the IMD mRNA level of calcified vascular tissue did not change when compared with controls. 33 In hypercholesterolemia rats, the expression of IMD and all its receptor components was increased in the aorta. 43 The inconsistent trend in aortic tissue may be attributed to different disease states and models. Overall, the changes in expression of IMD and its receptors in the Hcy-induced atherosclerosis calcification model suggest that IMD may have a protective role in atherosclerotic calcification.
Vascular calcification can be classified as medial calcification, intimal calcification, and valve calcification. Medial calcification is highly associated with chronic kidney disease, age, and diabetes, whereas intimal calcification is often seen in atherosclerotic lesions. 44 Accumulating evidence affirms that IMD is a potent endogenous cardiovascular protective peptide and extensively distributed throughout the body. 28 Whether IMD has a protective role in the development of atherosclerotic calcification remains unknown, although our previous studies and those of other colleagues have indicated that IMD can inhibit vascular medial calcification 32,33 and atherosclerosis. 29 -31,37 In this study, we found that IMD administration greatly ameliorated Hcy-accelerated atherosclerosis and atherosclerotic calcification in vivo. In addition, IMD pretreatment could reduce Hcy-promoted VSMC calcification in vitro.
The ERS is initially an adaptive response and maintain ER homeostasis through the unfolded protein response. The unfolded protein response involves 3 distinct signal transduction pathways mediated by the molecules PERK, IRE1, and ATF6. The 3 pathways can ameliorate the accumulation of unfolded protein by increasing the ER-resident chaperones, inhibiting protein translation, and promoting the degradation of unfolded proteins. However, when ERS is prolonged or severe, it initiates apoptotic cell death signaling. 12
Compelling evidence indicates that ERS is involved in the development and progression of atherosclerosis and vascular calcification. Endoplasmic reticulum stress can occur at all stages of atherosclerotic lesion development 45 and is associated with the acute coronary syndrome. 46 A series of reports also indicated that ERS contributes to vascular calcification, and inhibition of ERS retards vascular calcification. 13 -15 Here, we found that Hcy treatment promoted the protein expression of ERS markers in atherosclerotic lesions, which was reduced by both IMD and PBA administration. In light of the evidence that PBA, a chemical chaperone, known as an ERS inhibitor, could ameliorate Hcy-promoted atherosclerotic calcification, IMD may prevent Hcy-promoted atherosclerotic calcification by inhibiting ERS. However, the direct evidence that IMD prevents atherosclerotic calcification via its anti-ERS effect and the signal transduction pathway mainly regulated by IMD need further study. In addition, previous study reported that IMD could prevent the progression of atherogenic diet-induced atherosclerosis in ApoE–/– mice through the improvement in circulating lipid profile. 30,37 In our present study, IMD had a significant lipid-lowering effect, which was consistent with previous study. 30,37 Therefore, the protective effect of IMD on atherosclerotic calcification might be in part through improving lipid profile.
Although initially considered a passive precipitation of crystals, atherosclerotic calcification was recently suggested to be an active cell-driven process similar to bone development. 9 Many osteogenic regulatory factors, such as OPN, OCN, BMP, and matrix Gla protein, are expressed in atherosclerotic lesions 6 -8 and osteochondrogenic differentiation of VSMCs plays an essential role during atherosclerotic calcification. 11 We found that Hcy treatment increased the protein expression of osteoblast-like cell markers such as ALP, BMP2, Runx2, and OPN in lesions, which could be reduced by both IMD and PBA treatment. We further confirmed both IMD and PBA could reverse the Hcy-accelerated osteoblast differentiation of VSMCs in vitro. Thus, IMD has a similar effect on inhibiting osteoblast differentiation of VSMCs as PBA. Moreover, IMD pretreatment also markedly reduced the Hcy-promoted protein levels of ERS markers in VSMCs, which was consistent with the change in osteoblastic marker levels. So the inhibitory effect of IMD on osteoblast differentiation may be mediated at least in part by inhibiting ERS.
Several studies provided evidence that atherosclerotic calcification is associated with an inflammatory status, and macrophages promote osteogenesis in atherosclerotic plaques and VSMC calcification by releasing pro-inflammatory cytokines. 18,47 Anti-inflammation treatment with statins prevented the progression of macrophage burden, osteogenesis, and calcification in an ApoE–/– mice model 18 and aortic stenosis in patients. 48 We found increased infiltration of macrophages and protein expression of MCP-1 in lesions with Hcy treatment, and administration of both IMD and PBA reduced the infiltration of macrophages and expression of MCP-1 in lesions. Our in vitro results further demonstrated that IMD pretreatment could decrease the Hcy-evoked inflammation response of macrophages, which was similar to the effect of PBA pretreatment. Along with an anti-inflammatory effect of IMD in macrophages, IMD also inhibited ERS induced by Hcy in macrophages. Thus, the anti-inflammatory effect of IMD could be attributed in part to its role in inhibiting ERS.
In summary, we provide experimental evidence that the novel endogenous bioactive peptide IMD could be a novel paracrine/autocrine factor that attenuates Hcy-promoted atherosclerotic calcification by inhibiting ERS, thus preventing the development of characteristic features of calcification in mice, including the osteoblast differentiation of VSMCs and inflammation response of macrophages. The cardiovascular protective peptide IMD, as an endogenous calcification inhibitor, may be a promising therapeutic strategy for treating atherosclerotic calcification in patients with hyperlipidemia having HHcy.
Supplemental Material
Supplemental Material, supplementary_figures-20190921 - Intermedin1-53 Ameliorates Homocysteine-Promoted Atherosclerotic Calcification by Inhibiting Endoplasmic Reticulum Stress
Supplemental Material, supplementary_figures-20190921 for Intermedin1-53 Ameliorates Homocysteine-Promoted Atherosclerotic Calcification by Inhibiting Endoplasmic Reticulum Stress by Jin-Ling Ren, Yue-Long Hou, Xian-Qiang Ni, Qing Zhu, Yao Chen, Lin-Shuang Zhang, Xin Liu, Chang-Ding Xue, Ning Wu, Yan-Rong Yu, Chao-Shu Tang, Zhong-Ping Ning, San-Bao Chai and Yong-Fen Qi in Journal of Cardiovascular Pharmacology and Therapeutics
Supplemental Material
Supplemental Material, Supplementary_Table_1._Primer_sequences_for_real-time_PCR - Intermedin1-53 Ameliorates Homocysteine-Promoted Atherosclerotic Calcification by Inhibiting Endoplasmic Reticulum Stress
Supplemental Material, Supplementary_Table_1._Primer_sequences_for_real-time_PCR for Intermedin1-53 Ameliorates Homocysteine-Promoted Atherosclerotic Calcification by Inhibiting Endoplasmic Reticulum Stress by Jin-Ling Ren, Yue-Long Hou, Xian-Qiang Ni, Qing Zhu, Yao Chen, Lin-Shuang Zhang, Xin Liu, Chang-Ding Xue, Ning Wu, Yan-Rong Yu, Chao-Shu Tang, Zhong-Ping Ning, San-Bao Chai and Yong-Fen Qi in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Author Contributions
J-.L.R. contributed to the conception and design, acquisition, analysis and interpretation of data, and drafted the manuscript. Y-.L.H., X-.Q.N., and Q.Z. contributed to the concept and design, and critically revised the manuscript. Y.C. and L-.S.Z. contributed to acquisition, analysis and interpretation of data, and critically revised the manuscript. X.L., C-.D.X., and N.W. contributed to the acquisition and analysis of data, and critically revised the manuscript. Y-.R.Y. and C-.S.T. contributed to the conception and design, and critically revised the manuscript. S-.B.C. and Y-.F.Q. contributed to the conception and design, interpretation of data and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant no. 31872790, 81670434, 91339203 to Y.F.Q), the Outstanding Clinical Discipline Project of Shanghai Pudong to Z.P.N. and the program of Shanghai Municipal Health Commission (ZK2019B25 to Z.P.N.).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.