
Abstract
Calcific aortic valve disease (CAVD) is a common cardiovascular disease in the elderly individuals associated with major morbidity and mortality. The process is characterized by multiple steps: lipid infiltration, inflammation, fibrosis, and calcification. Inhibitors of proprotein convertase subtilisin/kexin type 9 (PCSK9) represent a new therapeutic category of drugs for the treatment of dyslipidemia and atherosclerotic cardiovascular disease. Monoclonal antibodies of PCSK9 can result in substantial reductions in atherogenic lipoprotein cholesterol-carrying particles, especially lipoprotein(a), and thereby hold the potential for further reducing events associated with atherosclerotic cardiovascular disease. In this article, we reviewed the clinical and experimental studies in order to find the evidence of the involvement of PCSK9 in CAVD and the potential benefits of PCSK9 monoclonal antibodies in clinical therapeutics.
Keywords
Introduction
Calcific aortic valve disease (CAVD) is the most common form of valvular heart disease and the third leading cause of cardiovascular disease in the Western world, affecting about 26% of the population aged 65 years and above. 1 Several recent studies demonstrated that CAVD is a major contributor to cardiovascular morbidity and mortality both in the general and at-risk populations. 2 The increasing number of patients with age-related calcification is a problem in developed nations, and the incidence of CAVD is expected to continue to rise in the near future as global life expectancy increases. Therefore, the health problem and socioeconomic burden associated with CAVD are likely to increase substantially. 3
The CAVD seems to be a polyfactorial disease. Several traditional risk factors were associated with inflammation and calcification of the aortic valve, including age, diabetes mellitus, dyslipidemia, hypertension, and metabolic syndrome. 4 -8 However, the traditional risk factors mentioned above are fairly poor predictors of CAVD compared with the predictive power of these factors for vascular disease. 9 Clinical and experimental studies have been performed to evaluate histopathological changes of aortic valve fibrosis/calcification in recent years. All efforts aimed at finding medical therapy or therapeutic target that could prevent or decelerate the progression. Unfortunately, until now, all relevant pharmacological therapies have failed to alter the natural course of CAVD in humans. The sole definite treatment for severe aortic stenosis (AS) is surgical/percutaneous transcatheter aortic valve replacement with poor prognosis. 10 Therefore, it is still urgent to elucidate the etiology of calcification for developing drug therapies and prevention methods.
The CAVD was previously thought to be a passive, degenerative disease of aging. Recent landmark studies of aortic valves from humans and experimental animals have demonstrated that CAVD appears to be an active regulation process including chronic inflammation, lipoprotein deposition, renin–angiotensin system involvement, extracellular matrix remodeling, and activation of specific osteogenic signaling pathways and apoptosis. 11,12 Aortic valve interstitial cells (AVICs) could take 2 different pathways in the course of calcification: dystrophic calcification via cell death and osteogenic differentiation via a phenotypic change in AVICs. 12
Proprotein convertase subtilisin/kexin type 9 (PCSK9), primarily expressed in the liver, is a crucial protein in lipid metabolism by virtue of its pivotal role in the degradation of the low-density lipoprotein receptor (LDLR). 13 Moreover, experimental studies indicate that PCSK9 might accelerate atherosclerosis by promoting inflammation, endothelial dysfunction, and hypertension by mechanisms independent of the LDLR. Therefore, inhibition of PCSK9 is a promising therapeutic option. Several approaches to inhibit PCSK9 activity have been theoretically proposed, and monoclonal antibodies have been considered as the most promising approach and established a reduction in the incidence of cardiovascular events. 14,15 In this review, we mainly focus on the possible impact of PCSK9 and new PCSK9 inhibitors on CAVD.
Contribution of PCSK9 to CAVD by Modulating Lipid Levels
Earlier studies found that lipid disorders act synergistically with other risk factors to increase the prevalence of CAVD in general population. 16 The hypothesis that lipids play a role in the development of CAVD was further supported by the finding of diffused atherosclerotic lesions in the aortic leaflets of patients with familial hypercholesterolemia (FH). 17 The relation between CAVD and PCSK9, the new target for lowering lipids, is still uncertain.
The PCSK9 overexpression is proatherogenic mainly by reducing LDLR and resulting in increased serum cholesterol. Transgenic mice Tg(PCSK9) fed with a Western diet (WD) exhibited increased levels of plasma cholesterol, aortic cholesteryl esters, and severe aortic lesions, while knockout of PCSK9 gene significantly reduced aortic cholesteryl ester accumulation and protected WD-fed mice from atherosclerosis. 18 Interestingly, circulating LDL cholesterol (LDL-C) levels differed only slightly among these animals. It is still unclear whether the slightly higher LDL-C level fully accounts for the marked increase in plaque burden observed in these Tg(PCSK9) mice. The WD-fed Tg(PCSK9) mice develop aortic calcification as well. Pattern similarity with overexpression of LRP5 and Wnt was observed in the aorta of LDLR-deficient mice and Tg(PCSK9) mice, 19 so PCSK9 might be involved in calcification mechanisms in osteoblast-like cells.
Lipids and oxidized lipids were found in the vicinity of CAVD lesions. 20,21 Interestingly, PCSK9 is also detectable in human atherosclerotic plaques and is produced and secreted by smooth muscle cells (SMCs) of these plaques. Furthermore, PCSK9 secreted by human SMCs is functionally active and capable of reducing LDLR expression in macrophages, and SMC-conditioned media significantly affect LDLR expression in human macrophages as well. 22 It has been demonstrated that the LDLR of macrophage affects the rate of foam cell formation. Thus, it is possible that PCSK9 expressed by SMCs may decrease LDLR levels in macrophages preventing foam cell formation in the atherosclerotic plaques, and this positive action of PCSK9 may counteract the detrimental effect of plasma PCSK9 that reduces LDLR expression and promotes atherosclerotic lesion formation. 21 On the other hand, PCSK9 secreted by SMCs directly reduced LDLR expression and LDL-C uptake of macrophages, which might result in extracellular lipid accumulation and modification. 23
Despite promising animal experiments and nonrandomized human trials, 24 the prospective randomized controlled trials (RCTs) did not confirm the expected benefits from statin therapies (Table 1). 25 -28 Several possible reasons were proposed to interpret the conflicting results. Initially, although atherosclerosis and CAVD share some clinical and pathologic features at some point, CAVD is more complex and there may be overlapping nonatherosclerotic mechanisms involved such as shear forces and genetic factors. Furthermore, dyslipidemia was not the main risk factor in the RCT-enrolled patients. Additionally, patients with CAVD in advanced stages were included. Dysfunctional stress distribution and inflammation might already be self-perpetuating, and statins could not have a beneficial effect anymore in such stages. Finally, plaque stabilization in coronary atherosclerotic lesions is the major mechanism responsible for the beneficial effects of statins, but plaque rupture is not the cause of symptoms in CAVD and not a mechanism for CAVD progression. 29,30
Published Randomized Controlled Trials of Calcific Aortic Valve Disease Treated With Statins.

Abbreviations: AS, aortic stenosis; ASTRONOMER, Aortic Stenosis Progression Observation: Measuring Effects of Rosuvastatin; AVA, aortic valve area; AVC, aortic valve calcium; MG, mean transaortic pressure gradient; PG, peak transaortic pressure gradient; PV, peak aortic jet velocity; SALTIRE, Scottish Aortic Stenosis and Lipid Lowering Trial: Impact on Regression; SEAS, Simvastatin and Ezetimibe Aortic Stenosis; TASS, Tyrolean Aortic Stenosis Study.
The inhibition of PCSK9 and the maximally increasing LDLR as a result probably have synergistic advantages. The LDLR contributes to the clearance of atherogenic lipoproteins other than LDL-C, such as intermediate-density lipoproteins and remnant particles, the latter via APoE affinity. 23 Statin treatment did not improve CAVD development even in patients with no clinical signs of CAVD, therefore, if PCKS9 inhibition proves to be an important CAVD therapy eventually, it is likely to be independent of LDL-C reduction. Increased intermediate-density lipoproteins and remnant particle clearance may have real therapeutic benefits. 31
Lipoprotein(a) [Lp(a)], the preferential plasma lipoprotein carrier of oxidized phospholipids, is recognized as an independent risk factor for atherosclerotic cardiovascular disease and CAVD. Elevation in Lp(a) increases the risk for cardiovascular disease via its atherogenic LDL moiety and its prothrombotic, proinflammatory apolipoprotein(a) moiety. Growing evidence suggests that the genetically determined Lp(a) level was associated with CAVD. 32 -36 A genome-wide association study has identified a genetic variant (rs10455872SNP) in the LPA gene locus, which is the determinant factor of Lp(a) plasma levels. Elevated Lp(a) levels and corresponding genotypes are associated with increased risk of CAVD in the general population, with levels more than 90 mg/dL predicting a 3-fold increased risk. 33 Ker had found a clear association between plasma Lp(a) and calcification of the bicuspid aortic valve. 34 Lipoprotein-associated phospholipase A2 (Lp-PLA2) was highly expressed in aortic valves explanted from patients with CAVD. The Lp-PLA2 transported by the lipoproteins and/or released by macrophages hydrolyzes oxidized phospholipids carried by Lp(a) to generate lysophosphatidylcholine (LPC). As a powerful proinflammatory and atherogenic compound, LPC induces the expression of alkaline phosphatase and mineralization of AVIC cultures. Highly expressed Lp-PLA2 takes part in the mineralization of AVICs by the production of LPC eventually. 35,36 These evidence support that Lp(a) plays a causal role in CAVD, although there is currently no evidence that reducing Lp(a) will have therapeutic benefits. As described earlier, genetic variation in the LPA locus, mediated by Lp(a) levels, increases the risk of developing AS by >50%, and hence, it is worthy to explore the significance of Lp(a)-lowering therapies for CAVD.
The Lp(a) level is primarily genetically determined and relatively refractory to both lifestyle and drug intervention. Statins are ineffective in lowering the Lp(a) level. 37 Recent clinical trial showed that PCSK9 inhibition significantly reduces the incidence of major adverse cardiovascular events, in addition to reducing Lp(a) levels (Table 2). 15,38,39,45,46 Program to Reduce LDL-C and Cardiovascular Outcomes Following Inhibition of PCSK9 In Different Populations analysis confirmed that inhibition of PCSK9 yielded significant Lp(a) reductions in a dose-dependent manner. 40 However, the underlying molecular mechanism(s) of Lp(a) metabolism is not clear yet. The LDLR was considered to play a role in the uptake and degradation of Lp(a). But reports were controversial on the significance of the role of LDLR. The PCSK9 inhibitors reduce Lp(a) in patients with homozygous FH, despite their lack or dysfunction of LDLR. 41 Reblin et al identified that LDLR-deficient fibroblasts did not alter the catabolism of Lp(a). 42 Because Lp(a) clearance by hepatocytes appears to depend on very low-density lipoprotein receptor (VLDLR) expression, the modulation of VLDLR by PCSK9 appears to be of great interest. 43 The cellular uptake of Lp(a) was also dependent on clathrin-coated pits, and Lp(a) was targeted for lysosomal and not proteasomal degradation. 44 Thus, reducing PCSK9 expression or receptor binding activity may mediate the reduction in Lp(a) (Figure 1).
Published Phase 3 Randomized Controlled Trials of PCSK9 Monoclonal Antibody-Lowering Lp(a).

Abbreviations: GAUSS-2, The Goal Achievement after Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects study; Lp(a), lipoprotein(a); MENDEL-2, Monoclonal Antibody Against PCSK9 to Reduce Elevated LDL-C in Subjects Currently Not Receiving Drug Therapy for Easing Lipid Levels-2 trial; No., the number of all enrolled patients who were randomized; ODYSSEY LONG TERM, The Long-term Safety and Tolerability of Alirocumab in High Cardiovascular Risk Patients with Hypercholesterolemia Not Adequately Controlled with Their Lipid Modifying Therapy study; PCSK9, proprotein convertase subtilisin/kexin type 9; RUTHERFORD-2, The Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder-2 study; TESLA, The Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities study.
a P > .05.
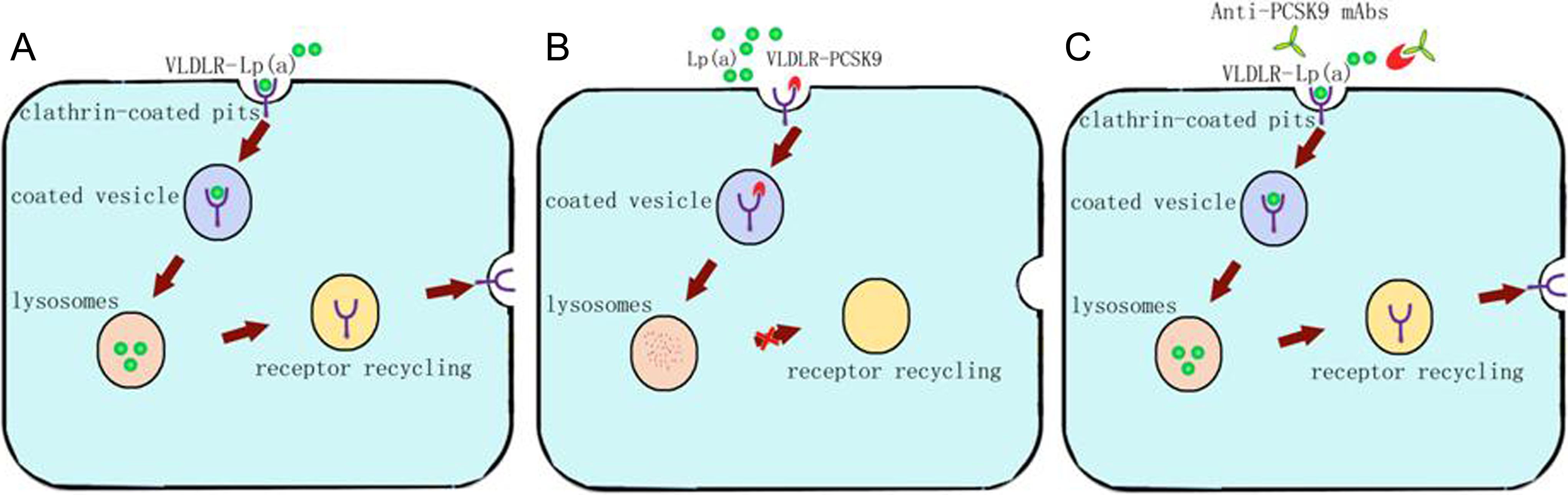
Possible mechanism of Lp(a) degradation through VLDLR and theoretical benefits of PCSK9 monoclonal antibodies. Catabolism of Lp(a) by VLDLR. The mechanism of Lp(a) degradation is not clear. A, Lipoprotein(a) may bind to VLDLR and degrade in lysosomes. B, Proprotein convertase subtilisin/kexin type 9 is involved in VLDLR recycling and mediate the reduction in Lp(a). C, Proprotein convertase subtilisin/kexin type 9 monoclonal antibodies yield significant Lp(a) reductions. Lp(a) indicates lipoprotein(a); PCSK9, proprotein convertase subtilisin/kexin type 9; VLDLR, very low-density lipoprotein receptor.
Those findings provide an interpretation of the therapeutic potential of PCSK9 inhibition in effectively lowering Lp(a) levels. It remains unknown whether lowering Lp(a) will yield clinical benefit in patients with CAVD. Nevertheless, the strong epidemiologic and genetic evidence suggest that Lp(a) is an independent causal risk factor for CAVD. Reducing Lp(a) levels via PCSK9 inhibition may be a potential target for medical therapy to reduce CAVD risk.
The correlation between PCSK9 and HDL cholesterol (HDL-C) has been investigated in several studies. 15,40,45,46 Plasma PCSK9 was positively correlated with plasma HDL-C. In spite of the mechanisms of the correlation between PCSK9 and HDL-C still need to be further explored, the more reasonable explanation is that ApoE-containing HDL particles may be cleared less efficiently because of LDLR degradation by PCSK9. 47 The HDL promotes the efflux of cholesterol from macrophages through adenosine triphosphate-binding cassette transporters. Moreover, HDL has antioxidative and anti-inflammatory properties. Clinical trials have confirmed that PCSK9 inhibition increased the levels of HDL-C and apolipoprotein A1. 48 -52 However, HDL does not appear to have any protective effect in the pathophysiology of CAVD. 53 No significant difference was found between patients with CAVD and controls in HDL-C levels, apolipoprotein A1 levels, lecithin–cholesterol acyltransferase activity, pre–β-HDL, HDL size, and 4 parameters of cholesterol efflux capacity including nonstimulated efflux, total efflux, ATP-binding cassette transporter A1 (ABCA1) efflux, and Scavenger receptor B1 (SR-B1) efflux. 54
Impact of PCSK9 on Inflammatory Pathways in CAVD
Inflammation is a prominent feature of CAVD. The chronic inflammatory infiltrates are associated with osteochondrogenic metaplasia and neovascularization in aortic valve. 55,56 Dense inflammatory infiltrate within the valves is associated with the active remodeling process, the severity of AS, and the hemodynamic progression rate. 57,58 Recently, Abdelbaky et al have found a direct evidence that early aortic valve inflammation may predispose to aortic valve sclerosis by fluorodeoxyglucose–positron emission tomography/computed tomography. 59 Inflammation is present in both early and advanced aortic valvular lesions. The early lesion of CAVD represents an active inflammatory process, which begins with disruption of valve endothelium, predominantly on the aortic side, followed by macrophages and T-lymphocyte infiltrations. Macrophage burden positively correlates with the extent of early cardiovascular calcification, and inflammation inversely correlates with bone mineralization. 60
In response to proinflammatory stimulation via activation of Toll-like receptors 2 and 4, the phenotype of myofibroblast VICs (AVICs) differentiates into osteoblast-like VICs (obVICs). The obVICs are characterized by an increased expression of osteoblastic differentiation markers, including bone morphogenetic protein 2 (BMP-2), the osteogenic transcription factor, Runx2, and an early mineralization marker, alkaline phosphatase. 61,62 Thus, understanding inflammatory signaling mechanisms may offer insight into selective abrogation of divergent calcific phenomena, and inhibition of inflammation might be valid only if targeted at initiating CAVD events. 11
Systemic inflammation is closely related to alterations in lipid metabolism. 63 -66 Although interaction between metabolic and inflammatory pathways is required for homeostasis, impaired cross talk among these pathways may also lead to metabolic dysregulation. Nicotine adenine dinucleotide (NAD)-dependent protein deacetylase sirtuin 1, a regulator of macrophage biology, was found to decrease the expression of resistin. This hormone leads to the accumulation of LDL-C in arterial walls and is positively correlated with CAVD in elderly patients. 2
Lipopolysaccharide (LPS) results in a marked increase in hepatic PCSK9 messenger RNA (mRNA) levels; similarly, zymosan, turpentine, and other treatments that induce inflammation also stimulate hepatic expression of PCSK9. The PCSK9 overexpression induced by inflammation leads to increased LDLR degradation and further increases circulating LDL-C levels. 67 Recently, Giunzioni et al found that expression of human PCSK9 in LPS-stimulated macrophages increased mRNA levels of the proinflammatory markers, tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β), and suppressed those of the anti-inflammatory markers, IL-10 and Arg1 in vitro. 68
The PCSK9 is upregulated by oxLDL stimulation in a dose-dependent manner in THP-1 (Human acute monocytic leukemia cell line)–derived macrophages. Small interfering RNA (siRNA)-mediated knockdown of PCSK9 suppresses the oxLDL-induced inflammatory cytokine expression. 69 Another proinflammatory mechanism might be gain-of-function mutant-type or wild-type PCSK9-induced degradation of ApoE-receptor 2, which maintains an anti-inflammatory phenotype in macrophages and mediates antiapoptotic signaling. 23,70 The circulating PCSK9 concentration correlated positively with high-sensitivity C-reactive protein levels besides total cholesterol and LDL-C. 71 C-reactive protein (CRP) was found in the fibrosa of human stenotic valves, and the presence of CRP in aortic stenotic valves could activate the classical complement system. 72
The TNF-α induces AVIC calcification through several mechanisms. Basically, TNF-α upregulates alkaline phosphate activity as a necessary component of vascular calcification in vitro. 73 The presence of TNF-α has also been linked to the activation of the receptor activator of nuclear factor kappa β (NF-κB) ligand, a key regulator of bone turnover and lymphocyte differentiation. 74 Berberine pretreatment reduced the expression of hepatic PCSK9 and then decreased the plasma TNF-α concentrations, 75 whereas PCSK9 siRNA significantly suppressed the expression and secretion of IL-1α, IL-6, and TNF-α in THP-1–derived macrophages. 68 Therefore, PCSK9 is involved in inflammation directly or indirectly.
Autotaxin (ATX) is a lysophospholipase
Taken together, PCSK9 directly increases inflammation of atherosclerotic lesion, and PCSK9 inhibitors may have therapeutic benefits.
Proprotein Convertase Subtilisin/Kexin Type 9 Might Be Involved in Apoptotic Cell Death in CAVD
Apoptosis is a strong promoter of ectopic valve mineralization. The TNF-related apoptosis-inducing ligand mediates the calcification of AVICs in culture through mechanisms involving apoptosis. 77 It is characteristically present within calcific aortic valves and binds to the death receptor 4 and death receptor 5, which are incidentally overexpressed by AVICs during mineralization and provoke programmed cell death through a caspase 8 pathway. 78 Oxidized phospholipids and Lp(a) would trigger apoptosis in endoplasmic reticulum-stressed macrophages through a mechanism requiring both CD36 and Toll-like receptor 2. New evidence indicated that apoptosis was implicated in LPC-induced AVIC mineralization. 35,79 Given that PCSK9 inhibitors decrease Lp(a) obviously, further studies would be needed to investigate the impact of PCSK9 on apoptotic cell death in CAVD.
Proprotein Convertase Subtilisin/Kexin Type 9 May Cause Valve Calcification Indirectly
Ectopic calcification in aortic valve is associated with considerable mortality and morbidity. Calcific nodule formation in CAVD has been linked to plasma calcium, parathyroid hormone, and renal disease. 71 Furthermore, there is strong support for the concept that CAVD is an active process mediated by ectopic osteoblastogenesis and that these osteoblast-like cells express markers similar to those found in skeletal osteoblasts. 10,55,80 Activation of pro-osteogenic signaling cascades is deemed to be a central mechanism contributing to the initiation and progression of CAVD. The process is thought to be initiated by cytokines released from infiltrating inflammatory cells and from apoptosis of AVICs. 81 However, because of aggravating osteoporosis, therapies that were designed to retard the progression of CAVD by means of inhibition of osteoblastogenesis, such as Peroxisome proliferator-activated receptor gamma (PPARγ) agonists, may not be helpful. 82
Patients with FH caused by PCSK9 gain-of-function mutation exhibit an age- and gene dosage–dependent increase in the incidence of CAVD. 83,84 The WD-fed C57BL/6 Tg(PCSK9) mice exhibited high cholesteryl esters and severe aortic atherosclerotic lesions while developed CAVD. 18,19 Therefore, it is reasonable to infer that PCSK9 might be involved in the interplay between lipids, inflammation, and apoptosis and cause aortic valve calcification eventually.
Conclusions and Future Perspectives
Calcific aortic valve disease contains a wide spectrum of clinical entities from aortic sclerosis to severe AS. Patients often remain asymptomatic for long periods. Early lesions of CAVD and atherosclerosis have many similarities, including lipid deposition and inflammation. Medication including statins, angiotensin-converting enzyme inhibitors (ACE-Is), and angiotensin II type 1 receptor blockers failed to work in prospective RCTs until now. 2,85 However, statins/ACE-Is may have a protective effect when administered at a time before a fixed lesion has developed. 85 In addition, more and more evidence support that elevated Lp(a) appears to be a key causal factor of CAVD. 86 We need to identify patients in earlier preobstructive stages and to search the optimal moment to start medication therapy and to find the key therapeutic targets for this chronic progressive disease.
The PCSK9 plays a vital role in the regulation of cholesterol homeostasis by increasing the degradation of hepatic LDLR. In clinical studies, inhibition of PCSK9, mainly by PCSK9 monoclonal antibody, potently reduced circulating LDL-C and LP(a) dramatically. Experimental studies proved that PCSK9 might accelerate atherosclerosis in an LDLR-dependent or LDLR-independent mechanism. These preclinical experimental data suggest that further research is needed to discern the potential beneficial therapeutic effects. There is an extensive cross talk between PCSK9 and CAVD; however, there is no study directly addressing this topic until now. Therefore, it is of interest to systematically investigate the role of PCSK9 for the pathogenesis of CAVD and to test the helpful impact of PCSK9 inhibition on CAVD. The PCSK9 monoclonal antibodies are unlikely to be used (due to limited availability and cost) to treat the large population with early CAVD. If a beneficial link between PCSK9 inhibition and prevention of CAVD is established, small-molecule inhibitors would probably be required for clinical realization. We are excited about the future of PCSK9 inhibitors research and about the opportunity to perhaps at last see clinical trials that will put to test the hypothesis on the role of PCSK9 in CAVD.
Footnotes
Author Contributions
W. Wang contributed to conception, contributed to analysis, drafted the manuscript, and critically revised the manuscript. C. Liu contributed to design and critically revised the manuscript. H. Cong contributed to conception and design, critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Tianjin Municipal Science and Technology Commission (No. 2ZCZDSY03200) and China Postdoctoral Science Foundation (No. 2015M581308).