
Abstract
In the setting of acute myocardial infarction (AMI), adverse myocardial remodeling (AMR) has been universally regarded as an early-onset phenomenon generally arising within the first few weeks (usually within days in the infarct zone) following myocardial injury. On the other hand, onset of cardiac morphological changes in this setting may potentially extend far beyond this time frame (usually beyond several months after the index AMI), suggesting a prolonged latent period in certain cases. In clinical practice, this delayed form of post-AMI remodeling, namely late AMR, has emerged as an interesting and underrecognized phenomenon with poorly understood mechanisms. Notably, systemic inflammation and associated growth factors seem to play a pivotal role in this setting. Accordingly, the present article primarily aims to discuss potential mechanisms and clinical implications of late AMR (in a comparative manner with its classical early counterpart) among AMI survivors along with a particular emphasis on potential benefits of certain anti-inflammatory strategies in this setting.
Keywords
Introduction
In clinical practice, adverse myocardial remodeling (AMR) has been considered as an ominous finding of acute myocardial infarction (AMI) 1 –6 and appears to be characterized by a variety of gradual and time-dependent changes in left ventricular (LV) morphology including infarct expansion and chamber dilatation along with compensatory hypertrophy of noninfarcted (remote) myocardial segments (with increments in end-systolic and diastolic volumes ultimately leading to systolic dysfunction) as visualized on certain imaging modalities including cardiac magnetic resonance imaging (MRI) and echocardiography. 1,3 –6
Temporally, classical AMR in the setting of AMI is a relatively early-onset phenomenon usually emerging within the first few weeks (usually within days in the infarct zone) or occasionally within the first few months after the index event in a substantial portion of AMI survivors and appears to be strongly dependent on the magnitude of the initial infarct size. 1,4 –6 Importantly, LV systolic functions as measured with left ventricular ejection fraction (LVEF) value might drastically change in the early post-AMI setting possibly as a consequence of ongoing early remodeling process. Accordingly, current guidelines recommend the evaluation of LVEF at 3 months following primary percutaneous coronary intervention for clinical decision-making for implantable cardioverter-defibrillator therapy among AMI survivors. 7 This might possibly imply that classical AMR arising after AMI, despite its significant individual variation, is generally expected to be fully established at 3 months (at the latest). Therefore, we deem it more appropriate to term this widely recognized and classical form of myocardial remodeling as “early AMR” in the post-AMI setting.
On the other hand, onset of initial morphological alterations on imaging modalities may extend beyond several months or even a year, 4 suggesting a prolonged and variable latent period in certain settings. This particular form of post-AMI remodeling with a delayed onset was previously termed as “late AMR,” and was suggested to arise after the first 3 months following the index AMI. 4 However, a couple of reports have exclusively regarded “late AMR” as a variety of morphological changes representing later stages of classical early AMR (characterized by compensatory hypertrophy and dysfunction involving remote myocardium and usually arising ≥1 month after AMI) rather than a distinct phenomenon with a de novo presentation. 8,9 Notably, these late changes were also suggested to serve as the fundamental determinants of prognosis among AMI survivors. 9
Surprisingly, there has been no clear-cut and uniform categorization of myocardial remodeling suggested so far based on temporal trends and characteristics along with no mention of late AMR as a separate clinical and pathophysiological entity in the post-AMI setting. However, late AMR should not be regarded simply as the late stage of classical early AMR, but as a late-onset phenomenon (with a significant latent period) with diverse mechanisms and implications as compared with its early counterpart in the setting of AMI (including its particular association with persistent systemic inflammation). On the other hand, macrostructural characteristics (and hence, morphological definitions) of early and late AMRs (including myocardial hypertrophy, chamber dilatation, volume expansion as demonstrated with various imaging modalities including cardiac MRI, echocardiogram) are generally similar. 4 On the other hand, it is well known that even though these macrostructural changes might, to some extent, contribute to systolic functions during the initial stages particularly in the setting of early AMR, they usually appear to be associated with the gradual evolution of progressive heart failure (HF) in the long term (particularly if left untreated). Importantly, a large portion of late remodelers might erroneously be labeled as nonremodelers in the early post-AMI setting and might potentially go unnoticed in clinical practice till they present with advanced HF. Figure 1 demonstrates a comparative time line regarding the evolution of early and late AMR. In this partly hypothesis-generating review, we aim to highlight mechanistic and clinical implications of late AMR (in a comparative manner with its early [classical] counterpart) among AMI survivors along with a particular emphasis on novel therapeutic options including certain anti-inflammatory strategies for the prevention of this phenomenon.

A comparative time line regarding the evolution of early and late adverse myocardial remodeling (AMR). Early changes: primarily characterized by infarct expansion and chamber dilatation usually within the first few days following AMI. Late changes: primarily characterized by myocyte hypertrophy and diffuse fibrosis predominantly involving remote myocardial segments and generally commencing within weeks (occasionally within months). * denotes the onset of morphological changes. ** denotes a significant and variable period without evident morphological changes in imaging modalities. + the onset might occasionally extend beyond a year. AMI, acute myocardial infarction; AMR, adverse myocardial remodeling; HF, heart failure.
Classical (Early) Myocardial Remodeling After AMI: Novel Insights Into Pathogenesis
Mechanistically, it is well known that evolution and severity of early AMR in the post-AMI setting appears to be determined by certain factors including extent of initial myocardial injury, magnitude of compensatory alterations in myocardial contraction pattern (including hyperkinesis in remote myocardial segments), and the degree of neurohormonal response (including arginine vasopressin system, renin–angiotensin–aldosterone system [RAAS], and adrenergic activation). 1,3,5 –17 Temporally, classical remodeling might be further categorized into 2 phases17: the early phase (within the first 3 days after AMI) primarily characterized by infarct expansion (leading to wall thinning and chamber dilatation), and the late phase (beyond 3 days) characterized by myocyte hypertrophy as well as diffuse fibrosis involving the infarcted and remote myocardial segments. 17 However, the abovementioned conventional insights have been already discussed in detail elsewhere 1,3,5 –17 and hence are not within the scope of this review. On the other hand, certain novel mechanistic aspects merit further mentioning in this setting.
Dysregulated Inflammation Response With Consequent Infarct Overhealing
In the current literature, even though inflammation and associated fibrotic response have been predominantly discussed in the setting of pathological conditions including HF, myocardial remodeling, sepsis, and so on, 17 –24 these entities mostly function as physiological repair mechanisms in response to tissue injury: In the setting of AMI, inflammation response within the infarcted myocardial tissue invariably serves as the incipient phase of the healing response. 3 During this phase, a variety of endogenous danger signaling molecules, namely danger-associated molecular patterns (DAMPs) including heat shock proteins, are activated. 3,25 These DAMPs then stimulate certain transmembrane receptors, namely Toll-like receptors (TLRs; a subgroup of pattern recognition receptors [PRRs]) located on various cells of the immune system including monocytes, macrophages, dendritic cells, neutrophils, and epithelial cells. 3,25 –27 Stimulation of TLRs then activates nuclear factor-κB, potentially associated with the induction of a particular cascade of inflammation comprising prointerleukine-18 (pro IL-18) and prointerleukine-1β (pro IL-1β). 3 Danger-associated molecular patterns might also have the potential to activate certain molecular platforms including inflammasomes and associated caspase-1 that largely accounts for the maturation of pro IL-18 and pro IL-1β into their active forms. 3 This cascade of events finally leads to the migration of certain inflammatory cells including neutrophils toward the infarct zone that contains high levels of pro-inflammatory cytokines including IL-18, IL-1β, tumor necrosis factor α (TNF-α), and certain interferons. 3
As a prelude to scar formation, neutrophils are then gradually replaced by anti-inflammatory macrophages (M2 phenotype), lymphocytes, natural killer cells, dendritic cells, and more importantly myofibroblasts usually accompanied by increasing levels of anti-inflammatory cytokines including interleukin 10 (IL-10) and transforming growth factor β (TGF-β; with fibrotic and matrix-enhancing characteristics 28 ) in the infarct zone toward the repair (healing) phase. 3,29 Transforming growth factor β serves as the central mediator in the postinfarction healing phase and exerts its matrix-enhancing actions largely through Smad3 signaling. 21 Moreover, profibrotic actions of certain hormones including aldosterone and angiotensine-2 are, to some extent, mediated through TGF-β/Smad cascade as well 21 in this setting. Of note, myofibroblasts, which do not exist in normal myocardium, are generally originated from fibroblasts in response to TGF-β and other growth factors including platelet-derived growth factor along with the impact of neutrophils. 29 Importantly, these newly emerging myofibroblasts promote fibril-forming collagen synthesis and hence play a pivotal role in physiological infarct healing through induction of replacement fibrosis within the infarct tissue as well as reactive fibrosis in the remote myocardial segments. 29 However, this healing response may be strikingly abnormal in certain settings potentially contributing to AMR evolution in the post-AMI setting.
Within this context, a portion of AMI survivors with large infarcts might not incur AMR on follow-up. 3,6 In contrast, those with a limited infarct size might quickly and unexpectedly suffer AMR in certain settings. 3,6 Interestingly, the hypothesis of “dysregulated inflammation response” in response to tissue injury has been suggested in a recent paper. 3 Accordingly, individual variations (on a genetic and environmental basis) in inflammatory responses might help explain this AMR paradox encountered in certain patients following AMI. 3 The authors of this article particularly highlighted the pathophysiological relevance of augmented chronic persistent inflammation potentially associated with impaired healing response ultimately leading to AMR in certain AMI patients even with a limited infarct size. 3 Of note, even though this phenomenon might help unravel the mechanisms underlying unexpected occurrence of AMR in the setting of limited infarcts, 3 it might not fully explain the absence of remodeling in the setting of an extensive infarct in some AMI cases. Importantly, individual variations in neurohormonal responses (including certain genetic polymorphisms of β receptors and angiotensin converting enzyme) were previously documented and were suggested to have important clinical implications (remodeling, recovery of systolic functions) in the setting of HF. 22 These polymorphisms, on top of dysregulated inflammation and infarct overhealing, might also account for this AMR paradox in the post-AMI setting as well. However, this notion should be established through clinical studies. Taken together, it seems likely that an overhealing response within the infarct tissue largely due to the exaggeration of the abovementioned physiological regeneration pathways and possibly triggered by unknown mechanisms 3 potentially expedites the evolution of early AMR in certain settings among AMI survivors.
Notch Signaling
In the recent years, Notch signaling (mediated through the interaction of certain receptors [Notch 1-4] and ligands [jagged 1 and 2, delta-like (D) 2, 3 and 4] leading to proteolytic release of intracellular active mediator, namely Notch intracellular domain that primarily accounts for nuclear transcription of target genes during fetal cardiac development) has been suggested as an important modulator of inflammation and profibrotic response both in the early and in the late phases of classical post-AMI remodeling. 9 Moreover, Notch signaling appears to be significantly activated following AMI and serves as an important communication network between infarcted segments and remote myocardium (ultimately leading to a shift toward an embryonic phenotype within the remote segments characterized by coexisting apoptosis and hypertrophy along with emerging embryonic organelles). 9 On the other hand, Notch signaling was also shown to activate certain antiapoptotic genes, control cardiac hypertrophy along with promotion of cardiac regeneration, and improve endothelial functions. Certain congenital diseases including LV noncompaction are well known to be associated with mutations in Notch signaling. 9 Accordingly, individual variations in this signaling system might potentially determine the severity of early AMR as well. However, clinical relevance and therapeutic implications of Notch signaling in AMI survivors yet remain to be established. Taken together, early AMR may be considered as a quite complex and multifaceted phenomenon possibly with an individual variation largely determined by genetic and environmental factors. 3,9
In general, AMI survivors appear to be categorized into 3 subgroups according to the AMR status and its temporal pattern: nonremodelers, early remodelers, and late remodelers. 4 Among these, late remodelers, as described below, might be labeled as an underrecognized subgroup with unique features (including its stronger association with persistent systemic inflammation and certain profibrotic factors 1,4,23,24 etc) potentially suggesting important diagnostic, prognostic, and therapeutic implications.
Late Myocardial Remodeling After AMI: A Unique Phenomenon With Distinct Implications
Even though the ultimate pathological and macrostructural characteristics of early and late AMRs (as demonstrated with various imaging modalities including cardiac MRI, echocardiogram) are usually similar, 4 there potentially exist significant diversities between these 2 phenomenon with regard to their pathophysiological and clinical implications (Table 1). Accordingly, late AMR harbors a variety of unique pathophysiological and clinical characteristics.
Summary of Comparison Between Early and Late AMR After AMI.
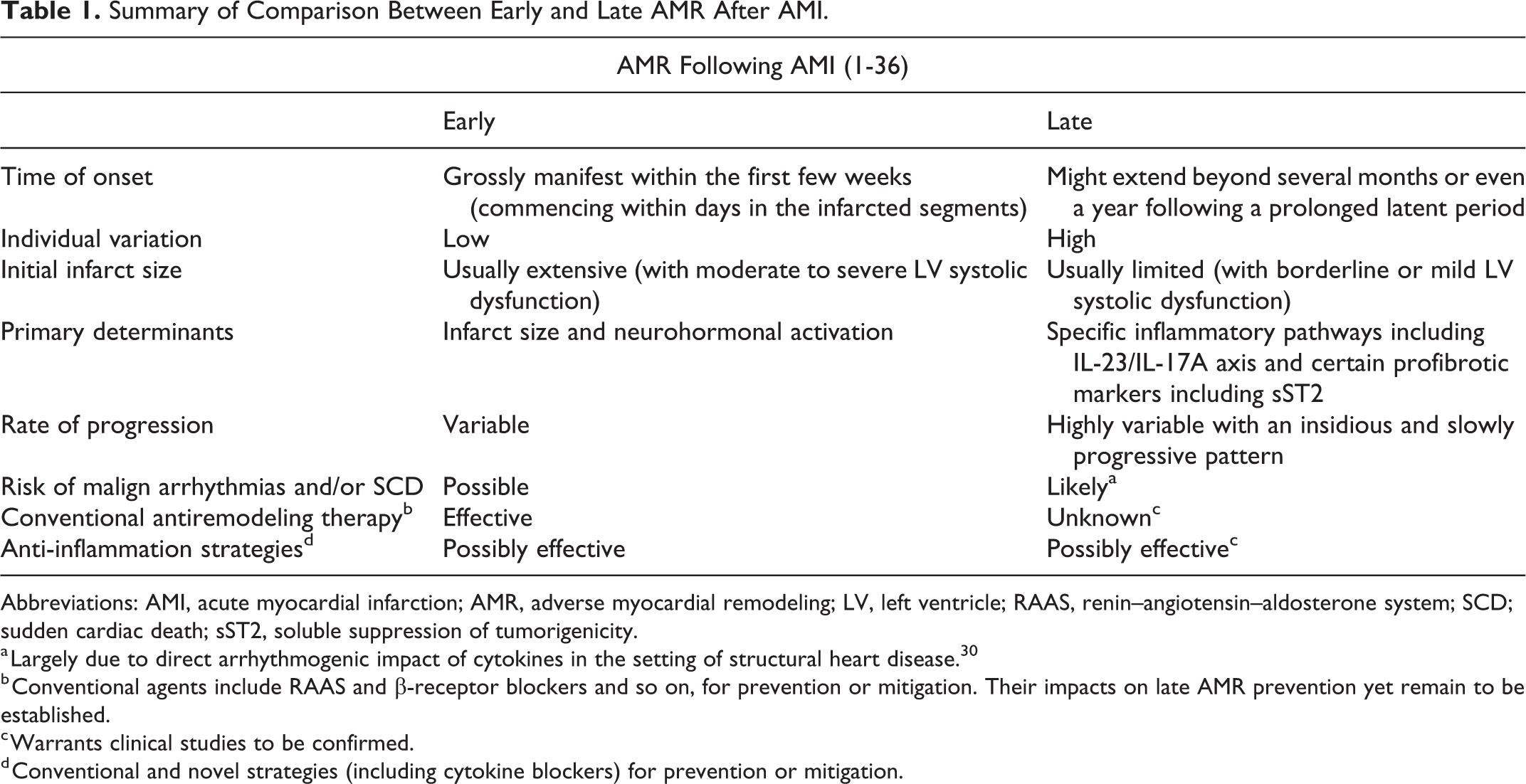
Abbreviations: AMI, acute myocardial infarction; AMR, adverse myocardial remodeling; LV, left ventricle; RAAS, renin–angiotensin–aldosterone system; SCD; sudden cardiac death; sST2, soluble suppression of tumorigenicity.
a Largely due to direct arrhythmogenic impact of cytokines in the setting of structural heart disease. 30
b Conventional agents include RAAS and β-receptor blockers and so on, for prevention or mitigation. Their impacts on late AMR prevention yet remain to be established.
c Warrants clinical studies to be confirmed.
d Conventional and novel strategies (including cytokine blockers) for prevention or mitigation.
Extent of Initial Myocardial Injury
In clinical practice, late remodelers generally suffer a borderline or mild LV dysfunction as compared with early remodelers who usually present with extensive infarcts: Accordingly, patients with AMI with an early AMR (presenting within the first 3 months) were previously demonstrated to have a significantly greater infarct size (as measured with MRI) in comparison to those with a late AMR (emerging after 3 months). 4 This finding 4 might imply that large infarcts and associated neurohormonal activation 1 might be expected to induce morphological changes already starting in the early postinfarction phase leading to early rather than late AMR and hence might possibly have little or no impact on the evolution of late AMR that emerges after a significant latent period. In other terms, significant neurohormonal activation might not be considered as a characteristic finding among late remodelers unless they progress to overt HF in the long term. In contrast, as described below, a variety of humoral factors might have a central role in the evolution of late AMR. Importantly, patients with large infarcts who are initially deemed as nonremodelers may occasionally undergo late AMR, potentially suggesting close monitoring of these patients even long after the index event particularly in the presence of persistent systemic inflammation. Since the infarct size appears to be limited in an overwhelming majority of late remodelers, 4 characteristic changes in early remodeling immediately involving the infarcted segments (infarct expansion, wall thinning) 17 appear to be quite unusual in these patients.
Mechanistic Basis: A Stronger Association With Persistent Systemic Inflammation and Associated Growth Factors
Persistent inflammation stemming from remote myocardium: An important determinant of late AMR evolution
Inflammation in the setting of AMI may be termed as “sterile inflammation” 18 and has 2 potential myocardial sources: necrotic tissue (as mentioned in the early AMR section) and remote myocardial segments. After infarct healing, myocardial tissue might still serve as a fundamental source of systemic inflammation largely due to the ongoing myocardial wall stress (myocardial stretch and hemodynamic load) that accounts for the release of certain cytokines including TNF-α, IL-18, IL-6, and IL-1β. 18,19 Genetic polymorphisms associated with exaggerated production and release of these cytokines might significantly contribute to post-AMI remodeling 3 in the setting of enhanced myocardial wall stress. Similarly, soluble suppression of tumorigenicity (sST2) has drawn a substantial interest as a strong profibrotic substance due to its neutralizing impact on IL-33 (a potent antifibrotic and antihypertrophic cytokine) and was reported to rise early after AMI in correlation with infarct size. 17,23,31 This also suggests enhanced myocardial wall stress as the fundamental trigger of sST2 production in the setting of AMI. 23 Interestingly, a recent study has suggested the particular value of increased sST2 levels (at 3 months after AMI) in predicting late AMR (vs nonremodelers) in patients with AMI. 4 On the other hand, this study did not elaborate on the issue of whether sST2 just served as an innocent bystander or directly triggered late AMR in post-AMI patients. 4 Nevertheless, it may be suggested that ST2 serves as an important marker of late AMR evolution on top of certain cytokines. 3,4,18 –21,32
More recently, a crucial inflammation pathway, namely IL-23/IL-17A axis, along with associated specific cells including gamma-delta T (γδT) cells were shown to have a particular association with late AMR evolution in experimental studies. 33,34 Moreover, this inflammation axis was previously shown to be implicated in ischemia–reperfusion injury 35 and postmyocarditis remodeling as well. 36 These experimental studies 33,34 suggest IL-17A as the central mediator predominantly produced by γδT cells through the stimulation of upstream regulatory molecules including IL-23, IL-1 β, and certain receptors including TLRs (by DAMPs) located on these cells. Of note, myocardial infiltration by γδT and associated IL-17A expression emerges in the later stages after infarct healing and hence are not involved in the acute inflammation stage in the early post-AMI setting. 33 In contrast, IL-17A was previously shown to have strong and late-onset apoptotic and profibrotic impacts on remote myocardial segments along with the downstream induction of macrophages to express IL-1 β, TNF-α, IL-6, and certain matrix metalloproteinases (MMPs; including MMP-2 and MMP-9). 33,34 Within this context, cardiomyocyte apoptosis has been regarded as one of the cardinal signs of late AMR and is largely attributable to p38 mitogen-activated protein kinase (MAPK)-p53-Bax signaling associated with intrinsic apoptotic pathway utilizing endoplasmic reticulum and mitochondria in this setting. 33,34 Similarly, profibrotic impact of IL-17A was suggested to be attributable to direct fibroblast activation, activation of fibrogenic genes leading to overexpression of highly fibrogenic mediators including MMPs and IL-6. 33,36
It is noteworthy that widely recognized pro-inflammatory mediators appear to serve as either upstream or downstream signaling molecules (for instance, IL1-β serving as both upstream and downstream signaling mediator) in the IL-23/IL-17A axis. 33 –36 This might signify the potential association between pro-inflammatory mediators and late AMR evolution in humans (through IL-23/IL-17A axis), suggesting these mediators as therapeutic targets in this setting as well. On the other hand, even though these experimental studies (33,34) provide critical insights, they do not specifically elaborate on the issue of whether the term ‘late AMR’ particularly refers to a separate and de-novo phenomenon with distinct implications and characteristics (latent period, etc.). Therefore, further experimental and clinical studies are still warranted to substantiate these preliminary findings, and to fully establish the definition and characteristics of late AMR.
Extramyocardial inflammation: Coexisting inflammatory conditions and their substantial impact on late AMR
Among AMI survivors, systemic inflammation may manifest as a chronic low-grade phenomenon (namely parainflammation) due to certain metabolic as well as nonmetabolic causes including obesity, metabolic syndrome, diabetes mellitus (DM), smoking, excess caloric intake, and sedentary life style. 3,18,30,37 –40 On the other hand, moderate high-grade inflammation may be encountered in the setting of chronic infections, 3 gout, 27 and certain rheumatologic conditions including rheumatoid arthritis and cancer. 18,41 –44 Interestingly, chronic and persistent systemic inflammation might per se have the potential to induce certain inflammatory conditions including DM, metabolic syndrome, and cancer, 27,45 potentially creating a vicious cycle between systemic inflammation and certain inflammatory diseases. Accordingly, IL-1β might serve as a crucial mediator in the evolution of type-2 DM (through enhancement of insulin resistance) 27 and gastric carcinoma, 45 both of which might further augment systemic inflammation largely through oxidative pathways. Apart from detrimental impact of IL-1β, dysregulation in expression of certain adipokines and cytokines (mostly attributable to abundance of pro-inflammatory macrophages within the adipose tissue leading to increased TNF-α levels along with decreased adiponectin levels) appears to be strongly associated with insulin resistance in the setting of type 2 diabetes and obesity. 18 Moreover, IL-1β produced by pro-inflammatory macrophages within the adipose tissue potentially stimulates bone marrow progenitor cells to produce neutrophils and monocytes leading to further infiltration of adipose tissue by these macrophages. 18 This also suggests an existing positive loop among pro-inflammatory macrophages, IL-1β, and bone marrow progenitor cells potentially associated with perpetuation of insulin resistance in this setting. 18
Mechanistically, cytokine response in correlation with the severity of systemic inflammation serves as the fundamental trigger of profibrotic and mitogenic mediators in the setting of extramyocardial inflammatory states: Accordingly, sST2 (a proven marker of late AMR) 4 was previously demonstrated to increase in various systemic inflammatory conditions as part of counterbalancing anti-inflammatory mechanisms. 24 Moreover, as described in the previous section, the overexpression of IL-23/IL-17A axis 33,34 might possibly have a significant role in this setting. However, as highlighted previously, late AMR might not only be associated with overt and rampant inflammatory conditions characterized by significant levels of inflammation markers but might also be attributable to a variety of subtle inflammatory disorders, for instance, type 2 DM and obesity, the well-known epitomes of parainflammation, are primarily characterized by adiponectin deficiency (possibly associated with subtle increases in TNF-α levels) that has a significant potential to induce AMR and diastolic HF in the clinical setting. 18 Taken together, coexisting inflammatory states might possibly exert their detrimental effects largely through induction of certain profibrotic, hypertrophic, and apoptotic mediators along with their significant impact on adipose tissue hormones including adipokines in the setting of late AMR. 18,33,34,36
On the other hand, it seems plausible that the abovementioned inflammatory conditions might serve only as an adjunct to the pathogenesis of early AMR and hence might rarely be potent enough in isolation (in the absence of classical remodeling triggers including extensive infarcts) to account for the evolution of this phenomenon. In contrast, it may be suggested that systemic inflammation, regardless of other remodeling triggers, appears to be strongly and primarily associated with the genesis of late AMR that is generally preceded by a long latent period and potentially unmasked only after an incessant and prolonged duration of myocardial bombardment with inflammatory and profibrotic mediators. However, these notions currently remain speculative and warrant clinical studies to be confirmed. Moreover, systemic inflammation, even in the absence of AMI, has the potential to induce AMR and HF in the long term. 18 However, an existing AMI even with a limited infarct size significantly facilitates this pathological process usually presenting with late AMR in this setting. Taken together, it seems plausible that systemic inflammation and associated growth factors (particularly stemming from remote myocardium and extramyocardial sources) seem to play a potential causative role in the setting of late AMR. 4,23,33,34 Figure 2 demonstrates potential sources of inflammation and their relative contributions to the evolution of AMR subtypes in the setting of AMI.
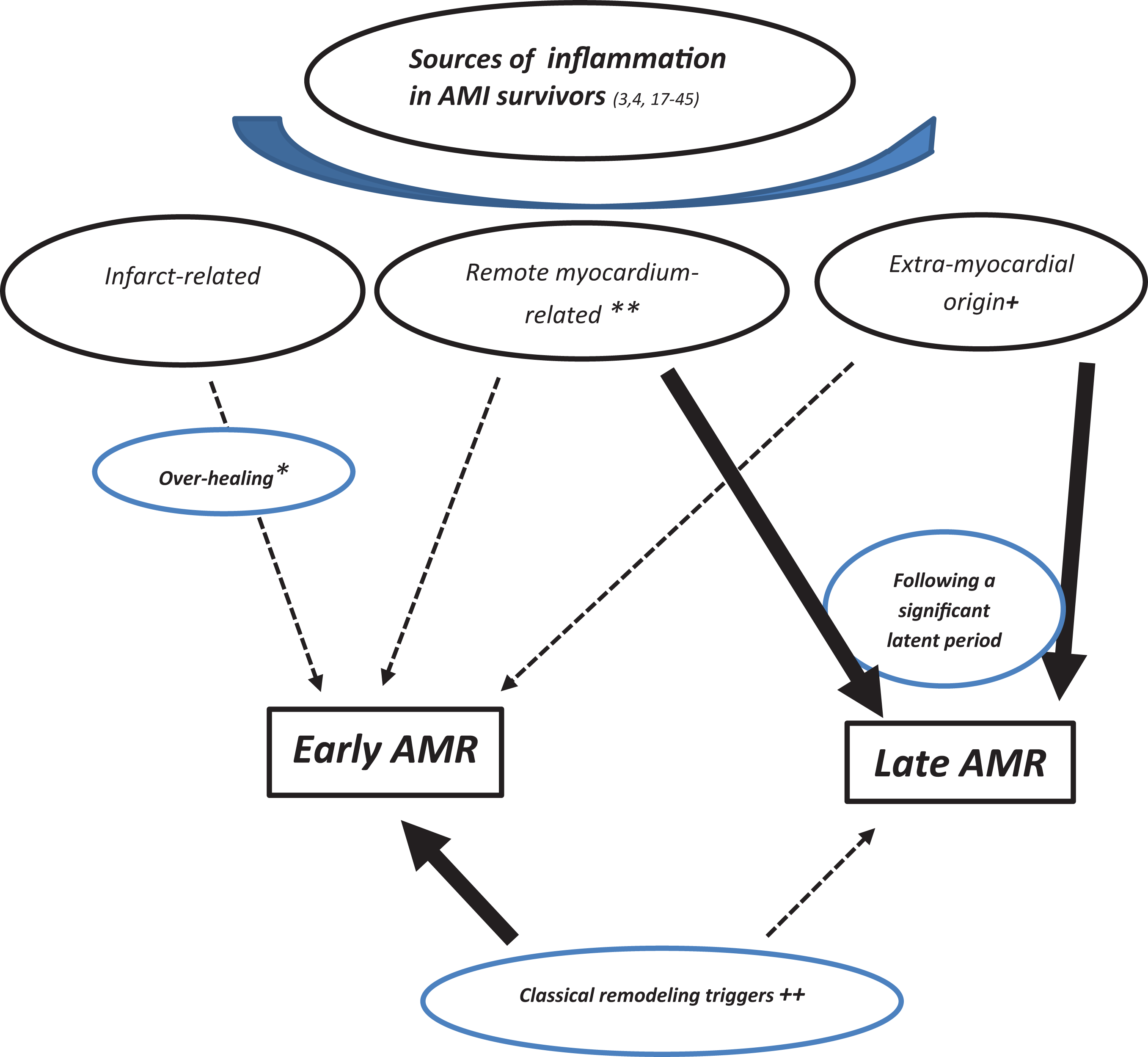
Sources of inflammation and their relative contributions to the evolution of adverse myocardial remodeling (AMR) subtypes in the setting of acute myocardial infarction (AMI). Black arrow: denotes substantial contribution. Dotted arrow: denotes weak contribution. * denotes exaggeration of physiological repair mechanisms due to substantial inflammation and profibrotic response within the infarct tissue leading to excessive scar formation in certain AMI survivors with a genetic or environmental predisposition. ** denotes attributable to pro-inflammatory mediators associated with enhanced wall stress and certain inflammatory pathways including IL-23/IL17A axis, and possibly exhibiting an individual variation largely determined by genetic and environmental factors. + denotes chronic infections, gout, rheumatologic conditions, cancer, and parainflammation including diabetes, metabolic syndrome, and so on. ++ denotes extensive infarcts, significant neurohormonal activation, and compensatory hyperkinesis. AMI, acute myocardial infarction; AMR, adverse myocardial remodeling.
Clinical Course
As mentioned earlier, late AMR is invariably preceded by a prolonged latent period of variable duration. After its emergence, the clinical course of this phenomenon may be more likely to have a more insidious and slowly progressive pattern with a significant individual variation largely determined by the status of circulating humoral factors including inflammation markers. Therefore, it seems imperative to combat ongoing inflammatory conditions even after emergence of late AMR. Importantly, a significant portion of late remodelers might initially be labeled as nonremodelers as there exists no evidence of AMR on serial imaging modalities (echocardiography, etc) performed within the first few months after the index AMI. In the setting of an uneventful early course following AMI, clinicians do not generally prefer to continue serial evaluation of LV morphology possibly overlooking the evolution of late AMR in certain participants. Therefore, late remodelers, even with full-blown HF, are generally less likely to receive guideline-directed HF therapy in a proper and timely manner potentially aggravating their prognosis in the clinical setting.
It seems noteworthy that even though certain metabolic conditions including obesity, type 2 DM, and metabolic syndrome have the potential to induce chronic and low-grade inflammation (namely parainflammation), they are widely regarded, by many clinicians, as sole endocrinological disorders rather than subtle inflammatory conditions with important implications in the setting of AMR and HF. Therefore, potential underdiagnosis and undertreatment of conditions with parainflammation (largely due to lack of information regarding detrimental impact of these conditions on emergence and perpetuation of late AMR) in late remodelers might further aggravate the prognosis in these patients. More importantly, since arrhythmogenic impact of systemic inflammation is well-documented particularly in the presence of structural heart disease, 30 late remodelers may be more likely to incur unexpected arrhythmic events and even sudden cardiac death long after the index AMI. On the other hand, it might be speculated that reverse remodeling (generally defined as a gradual and significant improvement in LV systolic functions along with a substantial diminution of LV volumes on follow-up 2 ) appears to be a more likely phenomenon among late as compared with early remodelers after management or mitigation of associated systemic inflammation possibly due to the relatively small infarct size in these patients. Interestingly, evolution of reverse remodeling might even be more likely in the setting of late AMR associated with parainflammation (particularly in the setting of obesity, type 2 DM, and metabolic syndrome in which inflammation-related adiponectin deficiency might serve as the fundamental trigger of AMR 18 ). This notion might also be indirectly substantiated by regression of LV hypertrophy in response to artificially created adiponectin overexpression in the experimental setting. 18
Therapeutic Implications: Potential Benefits of Conventional and Novel Anti-Inflammatory Strategies
Conventional agents including RAAS blockers 5 and β-blockers are well known to have a significant therapeutic value in the setting of classical early AMR. Furthermore, targeting systemic inflammation has also been suggested as a novel strategy with potential benefits in this setting. 3 On the other hand, based on a couple of experimental studies, 33,34,36 anti-inflammatory strategies might also be regarded as of potential value for the prevention or mitigation of late AMR. Moreover, since the primary trigger seems to be a state of systemic inflammation rather than infarct size in the setting of late AMR, the following strategies might be considered even more efficacious in this setting.
Management of coexisting inflammatory disease
In clinical practice, there might exist important therapeutic implications including proper recognition and specific management of aforementioned concomitant inflammatory conditions that might prevent or mitigate late AMR evolution in the post-AMI setting. On the other hand, it is well known that there exists no identifiable trigger of persistent systemic inflammation in a significant portion of patients in clinical practice. 30
Anticytokine therapies
Therapeutic implications of RAAS inhibition and β receptor blockade (with well-known inhibitory impacts on systemic inflammation, sST2 expression, 18,46,47 and other growth factors) in the setting of late AMR currently remain speculative and warrant clinical studies to be confirmed.
Conversely, antiinflammatory agents including cytokine blockers 32 might be of clinical value for the prevention of late AMR in the setting of persistent inflammation without an identifiable etiology. Within this context, beneficial impact of IL-1 antagonism on post-AMI cardiovascular outcomes through blockade of IL-1 receptors (with “anakinra”) as well as inhibition of circulating IL-1β (with “canakinumab”) in the recent reports 32,48 might have, to some extent, emerged due to the mitigation or prevention of late AMR among study participants. In experimental studies, IL-1β appeared to serve both as an upstream and downstream signaling mediator in the IL-23/IL-17A axis that was shown to play a pivotal role in late AMR evolution. 33,34 Therefore, IL-17A, IL-1β along with other widely recognized pro-inflammatory mediators that work as signaling mediators in IL-23/IL-17A axis might serve as important therapeutic targets for late AMR prevention 33,34 : within this context, even though soluble TNF-α (a downstream signaling mediator in IL-23/IL-17A axis) antagonist “etanercept” failed to yield any clinical benefit in patients with HF in terms of clinical status, hospitalization, and mortality, 3,49 it may also have a potential value for late AMR prevention. However, further studies on cytokine blockers are still warranted in this setting.
Targeting inflammasomes: A promising strategy
As described earlier, inflammasomes are considered as molecular platforms that primarily account for the maturation of certain cytokines including pro-IL-18, pro-IL-1β into their active forms through the activation of certain proteolytic enzymes including caspase-1. 3,27,45 On the other hand, stimulation of inflammasomes is generally mediated through activation of PRRs (TLRs, NOD-like receptors [NLRs], etc) of certain immune cells by host-derived (DAMP) or external antigens (pathogen-associated molecular patterns [PAMPs] associated with infectious agents). 3,27 The most spectacular subgroup of NLRs appears to be NLRP3 that is generally activated by a variety of DAMPs and PAMPs. 27 More specifically, NLRP3 activation was previously suggested to be attributable to adenosine triphosphate–mediated purinergic P2X7 receptor (P2X7R) stimulation leading to intracellular potassium (K+) reduction (due to K+ efflux). 45 Of note, NLRP3 actions might be potentiated in the presence of high oxidative stress. 45
Accordingly, activation of NLRP3 inflammasome (formerly termed as cryopyrin) by enhanced NLRP3 actions ultimately leads to the maturation of IL-1β (through caspase-1 actions) and was previously suggested to be associated with certain metabolic diseases with high inflammatory activity including gout and diabetes as well as certain pulmonary pathologies including silicosis and asbestosis. 27,45,50 –52 Moreover, gain-of-function mutations involving NLRP3 may manifest as a variety of genetic autoinflammatory diseases, namely cryopyrin-associated periodic syndromes presenting with inflammatory symptoms (urticaria, rash, etc). 45 Therefore, inflammasomes including NLRP3 inflammasome might be regarded as crucial determinants of systemic inflammation and important therapeutic targets in this setting. 27,45,50 –52 Even though there exists no direct inhibitor of NLRP3 inflammasome in the clinical setting, a variety of agents targeting upstream signaling including P2X7R inhibitors, antioxidants (N-acetylcysteine), sulfonylurea agents (inhibiting K+ channels), and downstream signaling including caspase-1inhibitors and IL-1β blockers have been tested and shown to yield promising results. 45 On the other hand, more detailed information on inflammasomes and their therapeutic implications may be found elsewhere 27,45 and are beyond the scope of this review.
Targeting specific immune cells
As described earlier, neutrophils have been considered as the initial inflammatory cells recruited within the infarct tissue and usually traverse the endothelium through a “CD11/CD18-integrin receptor” mediated process in this setting. 3 Following their migration, these cells primarily remove the cellular debris largely through phagocytosis as well as release of MMPs and reactive oxygen species and produce a variety of tissue factors potentially chemotactic to monocytes as well 3,21,53 ultimately preparing a proper milieu for the subsequent infarct healing. Of note, excess neutrophils within infarct tissue might theoretically account for additional tissue injury in the setting of AMI: Accordingly, inhibition of CD-integrin receptors (and hence hindering neutrophil migration) through the use of monoclonal antibodies was previously shown to diminish infarct size (an important determinant of early AMR 3,5,6 ) in experimental studies. 54 In contrast, clinical studies in this setting failed to demonstrate a significant benefit. 3,55,56
In an experimental study, 33 immunomodulatory agent FTY720 was shown to diminish myocardial infiltration by γδT cells (the cells reportedly accounting for late AMR evolution through IL-17 A actions) in the post-AMI setting partly by interfering with sphingosine-1 phosphate receptor signaling. More specifically, chemokine receptor 6 located on γδT cells and its ligand CCL20 within the injured myocardium might serve as important targets for the blockade of myocardial γδT cell recruitment in this setting. 33 However, these therapeutic options should be further tested in clinical studies.
Antifibrotic and antimitogenic strategies
As mentioned earlier, sST2 might have clinical implications in the setting of late AMR. 4 Therapeutically, blockade of sST2 with neutralizing antibodies has yielded promising results in certain noncardiovascular conditions. 57 However, implications of sST2 inhibition for the prevention of late AMR have yet remained to be established. More importantly, enhanced and unopposed effects of IL-33 as a consequence of sST2 inhibition might emerge as a therapeutic concern that needs to be clarified.
On the other hand, TGF-β might potentially arise as a more spectacular target in this setting largely due to its central role in infarct healing and multisignaling pathways (Smad signaling [mostly Smad3] and other non-Smad pathways [Rho GTPase; p38 MAPK; pathways). 21,58,59 Importantly, anti-TGF-β strategies have been extensively studied in a variety of diseases with severe fibrosis, including chemotherapy-induced pulmonary fibrosis and systemic sclerosis, and yielded promising results. 60,61 Therefore, it might be inferred that inhibition of TGF-β/Smad signaling (gene suppression, receptor blockade) might also serve as a promising target for the management of late AMR in the clinical setting. Accordingly, experimental studies testing the efficacy of anti-TGF strategies in the setting of cardiac fibrosis and remodeling have been quite promising. 62,63
Lastly, galectin-3 has also been suggested as an important target in this setting. 64 It is a well-known profibrotic mediator (β-galactoside-binding lectin) predominantly produced by the anti-inflammatory macrophages (M2 phenotype) and primarily accounts for infarct healing as well as AMR in the later stages of AMI largely through complex mechanisms including fibroblast to myofibroblast transformation. 64 In particular, experimental studies investigating the antifibrotic potency of certain galectin-3 inhibitors including antifibrotic tetrapeptide ac-SDKP, N-acetyllactosamine, and modified citrus pectin have yielded promising results. 64 Importantly, since anti-inflammatory as well as antifibrotic medications might potentially hamper infarct healing, 59 it seems more prudent to initiate these agents after complete healing of AMI 64 in an effort to obviate life-threatening complications including myocardial rupture and so on.
Modulators of Notch signaling
Activation of Notch signaling through myocardial delivery of Notch-1 pseudoligand or induction of Notch-1 transgenic expression was previously suggested to diminish myocardial fibrosis, apoptosis, and hypertrophic response associated with improvement in cardiac performance. 9,65 Interestingly, pro-inflammatory and necrotic cells were experimentally demonstrated to release a specific nuclear protein, namely high-mobility group box chromosomal protein. 9,66 Treatment with this protein was shown to promote cardiac repair and regeneration through induction of Notch signaling in a mouse experimental model. 66 Therefore, modulation of Notch signaling might have important therapeutic implications in the setting of late AMR. However, this notion should be confirmed through clinical studies. Table 2 presents promising anti-inflammatory strategies for the management of AMR in the post-AMI setting.
Promising Anti-Inflammation Strategies for the Management of AMR in the Post-AMI Setting.

Abbreviations: AMI, acute myocardial infarction; AMR, adverse myocardial remodeling; γδT, gamma-delta T.
Conclusion
In clinical practice, late AMR in the setting of AMI has risen as a poorly understood and underrecognized phenomenon that potentially harbors a variety of significant diversities with regard to its pathogenesis and clinical implications as compared with its early counterpart (despite their identical macrostructural characteristics). Since there exist limited data regarding this phenomenon in the current literature, certain implications mentioned in this article, to some extent, remain at a speculative level. Based on a few critical and enlightening studies as well as our clinical observations, it seems likely that the evolution of late AMR appears to be strongly associated with certain humoral factors including cytokines and associated growth factors rather than the major determinants of early AMR including infarct size and neurohormonal activation. Of note, there may exist a substantial variation in its severity as well as clinical course largely determined by the status of these humoral factors as well. As the name implies, the evolution of late AMR, as opposed to its early counterpart, is invariably preceded by a significant latent period during which a substantial portion of AMI survivors might readily and erroneously be labeled as a nonremodeler. This potentially warrants close and prolonged supervision of AMI survivors with persistent systemic inflammation along with the initiation of certain anti-inflammatory strategies in an effort to prevent potential occurrence of late AMR. In particular, certain therapeutic strategies (including inflammasome blockers, blockade of myocardial γδT cell recruitment, etc) might be quite promising for the management of AMR in the post-AMI setting. Accordingly, implications of this interesting phenomenon and its management strategies have yet remained to be established through further experimental and clinical studies.
Footnotes
Author Contributions
Kenan Yalta: Substantial writing, conception, design, literature search, acquisition, analysis and interpretation of literature data, preperation of tables and figures, major revision; Mehmet Birhan Yilmaz: Writing, literature search, analysis, interpretation, approval of revision; Tulin Yalta: Writing, literature search, analysis, interpretation, approval of revision; Orkide Palabiyik: Writing, literature search, approval of revision; Gokay Taylan: Writing, literature search, approval of revision; Cafer Zorkun: Writing, literature search, approval of revision.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.