
Abstract
Ranolazine has been found to prevent ventricular arrhythmias (VAs) during acute myocardial infarction (AMI). This study aimed to investigate its efficacy on VAs induced several days post-MI. For this purpose, 13 anesthetized rabbits underwent coronary artery ligation. Ten of these animals that survived AMI were reanesthetized 3 to 7 days later for electrophysiologic testing. An endocardial monophasic action potential combination catheter was placed in the right ventricle for simultaneous pacing and recording. Monophasic action potential duration, ventricular effective refractory period (VERP), and VAs induced by programmed stimulation were assessed. Measurements were performed during control pacing, and following an intravenous infusion of either a low-dose ranolazine (2.4 mg/kg, R1) or a higher dose ranolazine (4.8 mg/kg cumulative dose, R2). During control stimulation, 2 animals developed primary ventricular fibrillation (VF), 6 sustained ventricular tachycardia (sVT), and 2 nonsustained VT (nsVT). R1 did not prevent the appearance of VAs in any of the experiments; in contrast, it aggravated nsVT into sVT and complicated sVT termination in 2 of 6 animals. Sustained ventricular tachycardia cycle length and VERP were only slightly decreased after R1 (112 ± 5 vs 110 ± 6 ms and 101 ± 11 vs 98 ± 10 ms, respectively). R2 suppressed inducibility of control nsVT, VF, and sVT in 2 animals. In 4 animals with still inducible sVT, R2 significantly prolonged VT cycle length by 150 ± 23 ms (P < .01), and VERP by 120 ± 7 ms (P < .001) versus control. In conclusion, R2 exerted antiarrhythmic efficacy against subacute-MI VAs, whereas R1 rather aggravated than prevented these arrhythmias. Ventricular effective refractory period prolongation could partially explain the antiarrhythmic action of R2 in this rabbit model.
Keywords
Introduction
Ventricular tachyarrhythmias are a well-recognized and potentially lethal complication in patients with acute myocardial infarction (AMI). Up-to-day, there has been no established therapeutic strategy that effectively protects against such life-threatening arrhythmias. Management with classic antiarrhythmics has been shown to be inappropriate due to their unpredictable and largely proarrhythmic effects in the infarcted heart. 1
Focal electrical activity arising from abnormal automaticity or early and late afterdepolarizations following coronary artery occlusion is a common mechanism causing triggered activity and/or reentry under certain conditions. 2,3 Increased late sodium current (INaL) in the ischemically damaged myocardium may account as a mechanism for ectopic activity, generating after-potentials during repolarization due to sodium-overload mediated increased cytosolic calcium. 4 Although blockade of this current with ranolazine, a acetanilide and piperazine derivative that inhibits the INaL at therapeutic doses, has been suggested to be responsible for a substantial antiarrhythmic effect against ventricular arrhythmias (VAs) observed during AMI, 5 –7 its impact for VAs occurring several days postmyocardial infarction has not yet been explored.
Previous studies have shown that INaL blockade after relatively low doses of ranolazine reduces AMI-related VAs by depressing early after depolarizations (EADs). 8,9 In this context, although therapeutic doses of this agent have not been shown to significantly alter repolarization and refractoriness in the normal ventricle, 10 an impaired balance of the INaL and rapid component of the delayed rectifier potassium current (IKr) in infarcted myocardium might render it most vulnerable to arrhythmias. Specifically, a decrease of ventricular action potential duration due to a predominant INaL blockade may favor the conditions for reentry in ischemic areas.
Based on these eventually proarrhythmic properties of ranolazine when given at therapeutic doses after subacute MI, we aimed at investigating its electrophysiological effects at different dosing regimens, affecting primary the INaL or the INaL+IKr and examining the appearance of VAs induced by rapid pacing in rabbits 3 to 7 days after coronary artery occlusion.
Methods and Materials
Thirteen healthy, New Zealand, white rabbits of both sexes, weighing 2.5 to 3.5 kg were used for this study. The experimental protocol was approved by the Animal Care and Use Review Board at the University of Thessaly and conformed to all regulations for animal use.
Surgical Protocol
General preparation
All animals are premedicated intramuscularly with midazolam (1 mg/kg) plus ketamine (25-35 mg/kg) and anesthetized with thiopental sodium (30 mg/kg) administered slowly intravenously via the ear vein. Pancuronium 0.02 to 0.15 mg/kg was then intravenously injected to facilitate intubation. Mechanical ventilation was performed using room air mixed with oxygen via a volume-controlled ventilator (Harvard Apparatus, Holliston, Massachusetts) at a respiratory rate of 40 cycles/min and a stroke volume of 7 to 10 mL/kg. To maintain deep anesthesia, supplemental doses of thiopental sodium were given during the entire surgical procedure. The animals were placed in the supine position on an electrically heated pad and maintained at a core temperature of 38°C. Fluid-filled cannulas were inserted in the carotid artery to measure arterial pressure and in a peripheral vein for fluid and drugs infusion. A surface electrocardiogram (ECG) was continuously performed for detecting arrhythmias during the experiment.
Induction of acute MI
Surgical interventions were performed under sterile conditions. After a midline sternotomy, the pericardium was incised to enable exposition of the antero-lateral wall of left ventricle and the left anterior descending (LAD) coronary artery. In cases of a dominant LAD, the proximal segment of this artery was ligated; alternatively, the ramus interventricularis posterior (RIVP) of the left circumflex coronary artery was supplementary occluded at its distal third. The RIVP was approached by a soft left rotation of the heart.
Acute coronary artery occlusion was carried out by using a 3-0 monofilament polypropylene suture that was placed around the corresponding vessel including a part of the adjacent myocardium. During this procedure, the ECG limb, lead II, was continuously monitored using a NIHON KOHDEN RM-6000 polygraph amplifier system recorder (Tokyo, Japan). Myocardial ischemia was confirmed by ST-segment elevation on the ECG and by a sharp epicardial discoloration between the normally perfused and the ischemic myocardium (Figure 1).

Ligation of the left anterior descending (LAD) coronary artery resulting in a blue colored demarcation zone (arrows), separating the ischemic from the normal/healthy myocardium. The ligature knot at the left basal site of the heart denotes the location of the coronary artery occlusion.
Following acute infarction, the sternum was closed with nylon polyamide sutures No. 0 (3.5 metric), whereas the pericardial sac was left open. Postsurgical analgesia was ensured by subcutaneous administration of buprenorphine 0.01 to 0.05 mg/kg. Antibiotic prophylaxis with enrofloxacin 5 mg/kg intramuscularly twice a day was used for at least 2 days. Body temperature was maintained at 38°C during the first postoperative hours by an infrared heating lamp.
Electrophysiological Study
Recording system
All animals surviving the acute phase of MI (n = 10) were allowed to recover in a cage. After 3 to 7 days, each of these animals was subjected to electrophysiological testing under anesthesia and closed-chest conditions.
Peripheral veins were used for the infusion of fluids and drugs. Jugular veins were prepared for inserting and advancing the monophasic action potential (MAP) catheter to the right ventricle (Combination Catheter MAP-Stimulation 7F for in-vivo studies, Harvard Apparatus-HUGO SACHS ELEKTRONIK, March-Hugstetten, Germany). The electrode catheter was then gently pushed against the free wall of the ventricle until a stable MAP was obtained. To avoid direct-current (DC) drift, the MAP electrode was adequately soaked in saline before the insertion into the vein.
All signals were displayed on a multichannel monitor oscilloscope (NIHON KOHDEN RM-6000 polygraph amplifier system), equipped with a thermal array recorder (WS-682G). Monophasic action potential signals were amplified by a high input impedance, MAP DC-coupled isolated, differential preamplifier (AB-601 G). The frequency responses of the MAP recording system was from DC, at 5000 Hz.
Pacing protocol
Programmed and high-rate burst ventricular stimulation are performed to determine MAP duration (MAPD), VERP, and inducibility of sustained ventricular tachycardia or fibrillation. For that purpose, a Grass stimulator was used (Model S8800, Quincy, Massachusetts), delivering constant-current rectangular stimuli. Pacing pulses were of 2-ms duration at twice diastolic threshold intensity and 8 volts.
Ventricular MAP recordings were obtained during stable ventricular stimulation at 200 ms cycle length prior to and after drug administration. Monophasic action potential duration was assessed at 50% and 90% repolarization. Ventricular effective refractory period was determined using trains of 5 to 7 consecutive stimuli at 200 ms basic cycle length, followed by a premature stimulus at 10-ms decrements until no ventricular response appeared; then the extrastimulus cycle length was increased at 5-ms steps, until a local but not propagated ventricular response was recorded. Local responses were identified by the appearance of the MAP signal; propagation of a local response was determined by a QRS complex on surface ECG. The ERP was defined as the longest interval between the last basic stimulus and extrastimulus that failed to evoke a propagated ventricular response.
When programmed stimulation with a single extrastimulus did not elicit sustained ventricular tachyarrhythmias, high-frequency ventricular burst pacing of 80 to 120 ms cycle length was attempted. The goal of the tachypacing protocol was the induction and reproducibility of morphologically similar sustained ventricular tachycardia (sVT) during the control and after the bolus of a low-dose ranolazine (2.4 mg/kg, R1) or a higher dose ranolazine (4.8 mg/kg cumulative dose, R2). Pacing for evaluation of electrophysiologic variables and induction of arrhythmias was made during the first 10 to 15 min of either ranolazine dose regimen. When a DC shock was needed for restoration of sinus rhythm, continuation with the protocol was performed approximately 10 minutes after termination of arrhythmia.
Ranolazine dihydrochloride was purchased from SIGMA-ALDRICH (R6152, Darmstadt, Germany) and prepared separately before each experiment.
Arrhythmia definitions
Repetitive ventricular responses are defined runs of 3 to 6 beats occurring after cessation of pacing. Nonsustained ventricular tachycardia (nsVT) was defined as any tachycardia consisting of more than 6 ventricular complexes but terminated spontaneously within 30 seconds. Each hemodynamically stable VT composing of at least 100 uniform QRS complexes or lasting more than 30 seconds was defined as sVT, regardless of whether the tachycardia was terminated spontaneously or via overdrive pacing.
Proarrhythmic effects of the drug were considered when a nsVT during control stimulation showed spontaneous or overdrive pacing-induced transition into sVT or fibrillation. In addition, the inducibility of a faster sVT with or without degeneration to fibrillation was considered as a proarrhythmic action of the drug.
Study end points
Primary end points were reproducibility and changes in the course of inducible VAs after the first or second dose of ranolazine. Secondary targets of the study, included measurement of sustained ventricular tachycardia cycle length (sVT-CL), VERP, and MAPD at 50% and 90% repolarization.
Statistical Analysis
Results are expressed as mean (standard deviation [SD]). Paired t tests were used for comparing the electrophysiological variables before and after ranolazine in the same experimental animal. The statistical analysis was performed with Primer of Biostatistics, 7th edition, by Stanton A. Glantz (2017). Statistical significance determined using the Holm-Sidak method, with α set at .05.
Results
Of the 13 rabbits subjected to acute infarction, 2 died within the first hours after coronary occlusion and 1 animal succumbed to complications during intubation and anesthesia. The remaining 10 animals were effectively tested by programmed stimulation 3 to 7 days after AMI.
During control conditions, right ventricular (RV) pacing revealed primary ventricular fibrillation (VF) in 2 animals, sVT in 6 animals, and nsVT in 2 other animals (Figure 2). Ventricular fibrillation was terminated by DC shocks at 200 to 300 J and sVT by overdrive burst pacing. The lower dose of ranolazine (R1) did not prevent reproducibility of control VAs in any of the animals. At least 2 different VT forms were inducible in each animal during pacing, but only the reproducible ones were taken as reference for comparing the antiarrhythmic action of R1 and R2. Compared to R1, R2 suppressed reproducibility of control VF (2/2), nsVT (2/2), and sVT in 2 of 6 animals (Figure 2). In the remaining 4 of 6 animals with sVT, R2 did not prevent reinitiation of tachycardia.
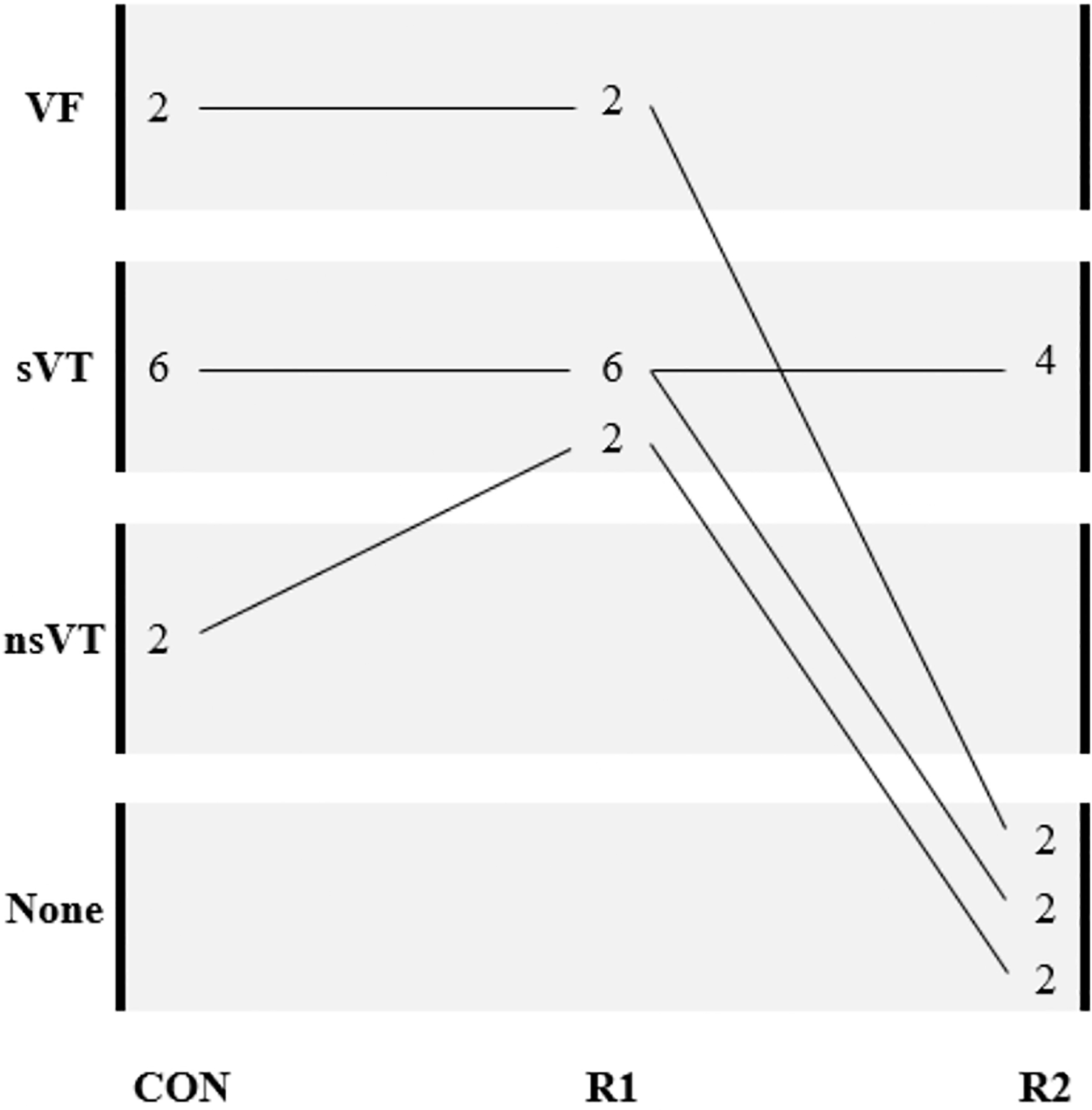
Schematic diagram illustrating the effects of the low-dose ranolazine (R1) and the high-dose ranolazine (R2) on rapid pacing induced ventricular tachyarrhythmias in subacutely infarcted rabbit hearts. VF indicates ventricular fibrillation; sVT, sustained ventricular tachycardia; nsVT, nonsustained ventricular tachycardia; None, no inducible arrhythmias. The animals are depicted by the numbers.
After R1, sVT-CL remained almost unchanged or was even slightly decreased (112 ± 5 vs 110 ± 6 ms, control vs R1, P = NS; Figure 3). In 2 of 6 rabbits which initially developed stable sVT, R1 facilitated transition of tachycardia to VF during overdrive burst pacing, which spontaneously reverted to sinus rhythm without the need of additional treatment. In the 2 animals displaying nsVT despite repetitive attempts of rapid ventricular pacing, R1 converted it to sVT that readily restored to sinus rhythm using overdrive pacing. R2 lengthened the median VT-CL to 150 ± 23 ms compared to control values (P < .01, n = 4; Figure 3). Figure 4 shows the effects of R1 and R2 on sVT reproducibility and cycle length in a representative experiment.
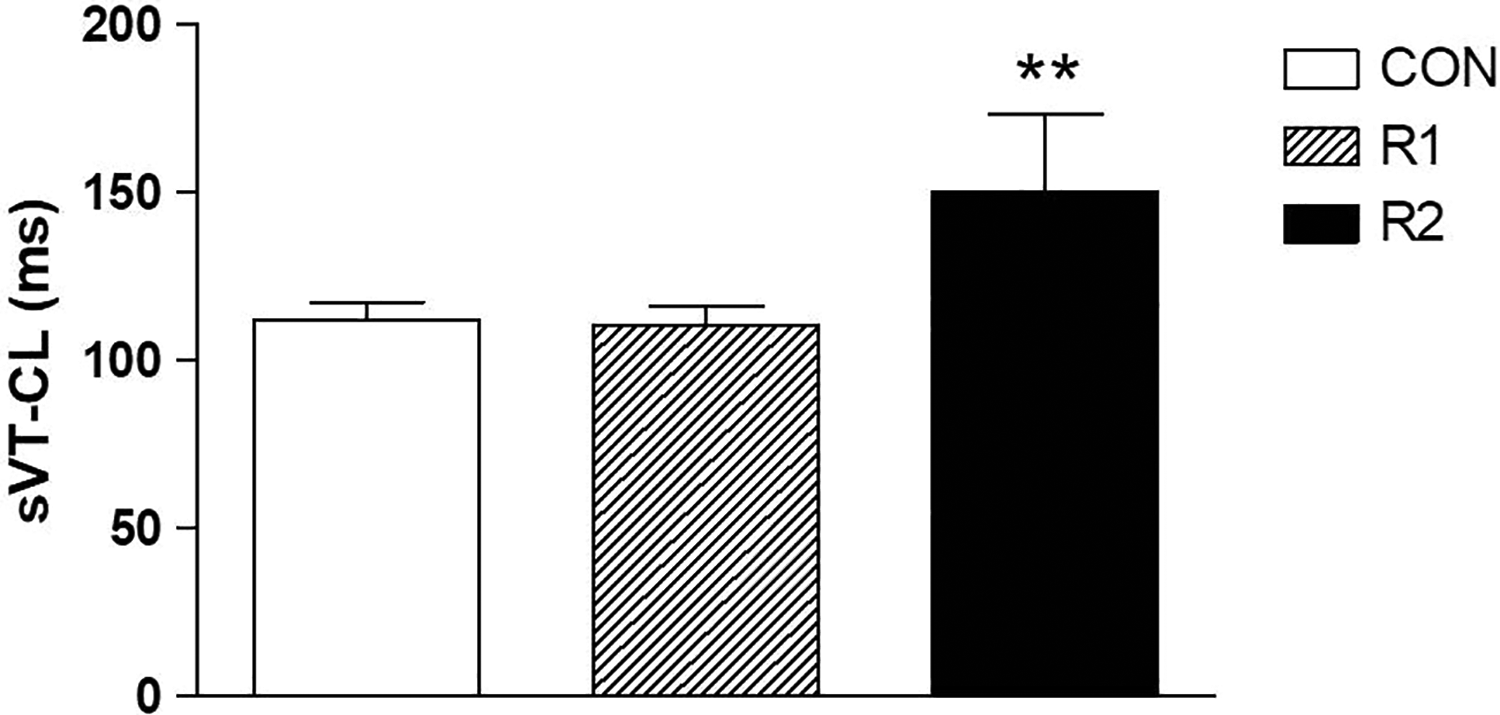
The low dose of ranolazine (R1) did not affect sustained ventricular tachycardia cycle length (sVT-CL) compared to control. In contrast, the high dose of ranolazine (R2) significantly prolonged sVT-CL in 4 of 6 animals (**P < .01, R2 vs Control).
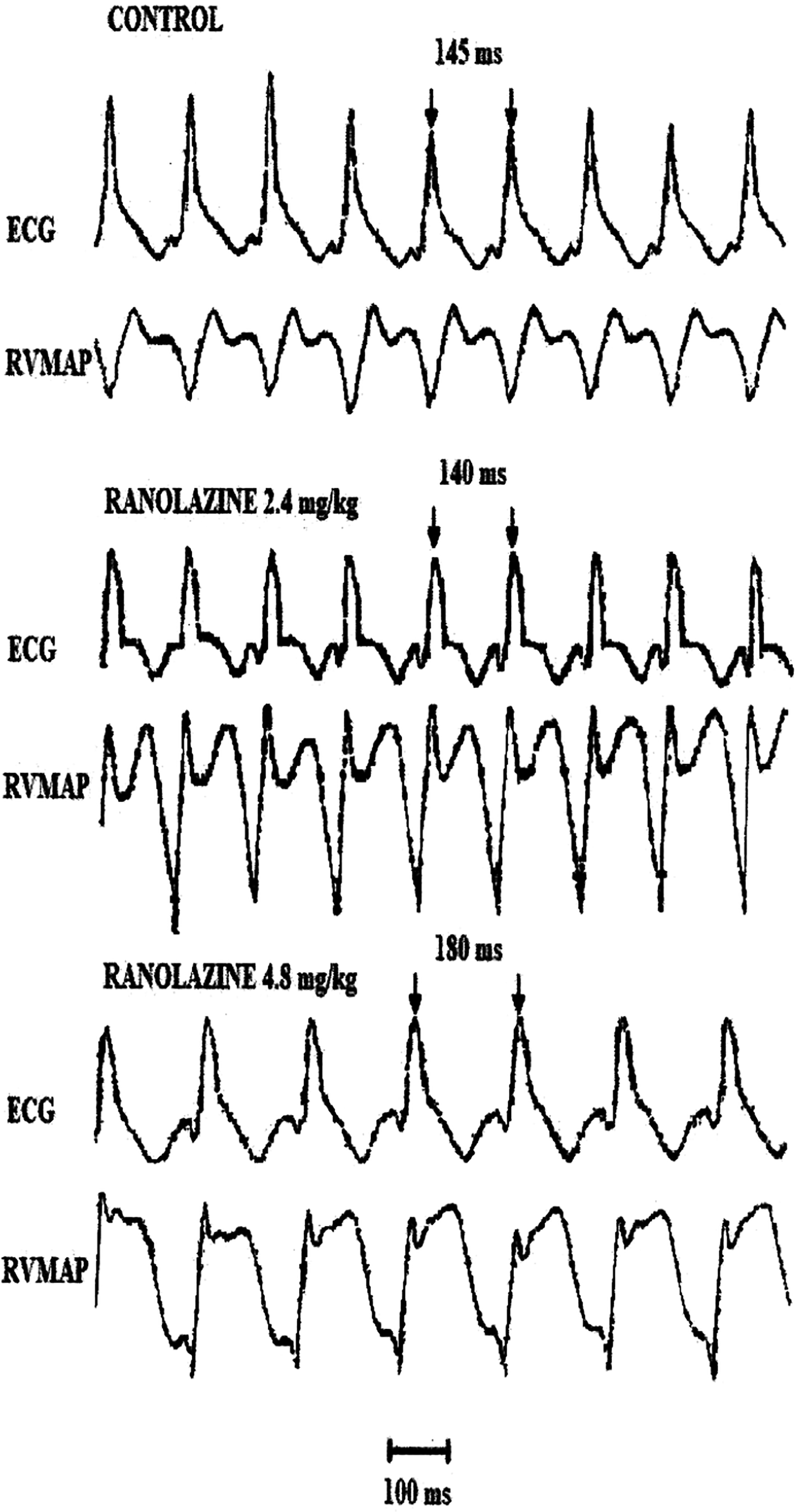
Representative data of sustained ventricular tachycardia (sVT) with rapid burst pacing after the low dose of ranolazine (2.4 mg/kg, R1) or the high dose of ranolazine (4.8 mg/kg, R2) in a rabbit, 5 days after AMI. R1 caused no significant changes on sVT-CL, whereas R2 markedly prolonged sVT-CL without suppressing its elicitation. The arrows indicate the cycle length of ventricular tachycardia during control and after R1 or R2. AMI indicates acute myocardial infarction; ECG, surface electrocardiogram; RVMAP, right ventricular monophasic action potential.
R1 tended to abbreviate the control VERP from 101 ± 11 to 98 ± 10 ms (P = NS), whereas after the second dose of ranolazine, refractoriness was significantly prolonged to 120 ± 7 ms (***P < .001; Figure 5).

Only the higher dose of ranolazine (R2) significantly prolonged ventricular effective refractory period (VERP; ***P < .001 R2 vs Control), whereas the low dose of ranolazine (R1) tended to shorten the VERP duration.
Monophasic action potential duration 50 was decreased after R1 from 71 ± 12 to 63 ± 6 ms (P = NS), and was prolonged to 76 ± 8 ms after R2 (R2 vs control, P = NS), however, these changes were not statistically significant. Monophasic action potential duration 90 was significantly reduced from 115 ± 9 to 106 ± 5 ms with R1 (P <.05), and lengthened again to 118 ± 9 ms after R2 administration (R2 vs control, P = NS; Figure 6).

Effect of first and second ranolazine dose (R1 and R2) on monophasic action potential duration (MAPD) determined at 50% and 90% repolarization. R1 shortened MAPD, whereas R2 prolonged MAPD determined during stable ventricular pacing (*P < .05, R2 vs Control).
Discussion
This study showed that only R2 exerted antiarrhythmic efficacy in the late post-MI period by suppressing the reproducibility of pacing-induced VAs or by slowing the sVT rate. This effect may partially be attributed to its VERP prolonging effect at the higher dose regimen. In contrast, the lower dose regimen did not change the course of these arrhythmias, but rather showed a tendency to deteriorate them into more protracted forms.
The main research question of this study was to examine whether the administration of ranolazine in patients several days after a MI, might raise their risk for VAs. This hypothesis was based on previous data showing that ranolazine at therapeutic concentrations inhibits exclusively the INaL (IC50 = 5.9 µM/L), 8 thereby may reduce ventricular repolarization and refractoriness. In the infarcted heart, even slight alterations in the action potential parameters might enhance the propensity to malignant VAs. For that reason, we assessed the effects of ranolazine on electrophysiological variables reflecting a predominant INaL or IKr blockade. Dhalla et al showed in rats that ranolazine administered at 2 mg/kg (low dose) and at 4 mg/kg (moderate dose) achieved plasma concentrations of maximum 5 and 13 µM within the first 5 minutes after bolus administration, respectively. 5 Experiments by Hale et al confirm this dose-concentration relationship in rabbits by showing that a 2 mg/kg bolus of ranolazine had reached concentrations of 3 to 5 µM, which are comparable to the therapeutic range used in humans. 11,12 These data indicate that our high-dose regimen of 4.8 mg/kg ranolazine possibly achieved concentrations capable of blocking the IKr (IC50 = 11.5 µM/L). 8 Investigators of the recently published RAID trial demonstrated a 30% reduction of malignant VAs using 1000 mg twice daily ranolazine orally (maximum dose of ranolazine) in implantable cardioverter-defibrillator patients at higher risk for developing VT/VF. 13
Although ranolazine has been reported to prevent VAs following AMI, yet there exist no data supporting its effectiveness during the subacute phase of MI. Ranolazine’s antiarrhythmic action during AMI has been shown to be mediated by inhibition of increased INaL rather than by IKr blockade. 5 –7,9 This can be explained by a reduction in INaL which in turn may decrease Na-dependent Ca2+ overload, suggesting as a potential mechanism of EADs. However, since the arrhythmogenic substrate during the late post-MI period is different and the mechanism of VAs is more likely linked to reentry, INaL blockade cannot effectively assist in preventing such arrhythmias solely on the basis of an EAD-suppressing mechanism. 14 The study by Morita et al 15 demonstrating a suppressing effect of selective INaL inhibition with ranolazine against hydrogen peroxide induced VF, predominantly refers to triggered activity initiated by EADs and not to conduction disturbances post MI. Ineffectiveness of the low ranolazine dose against inducible VAs in our experiments, corroborates the minor role of INaL inhibition in suppressing these arrhythmias. Conversely, if INaL > IKr inhibition, this would abbreviate the wavelength of potential reentrant pathways, thus speeding the rate of VT and worsening its course.
In comparison to the first ranolazine dose, R2 clearly showed an antiarrhythmic effect presumably due to a more potent IKr inhibition than R1. This might be consistent with a significant increase of VERP and sVT-CL in contrast to R1. It seems unlikely that these effects are attributable to changes in conduction, as the difference in VERP and MAPD90 values after R2 did not induce significant alterations in ventricular postrepolarization refractoriness. In addition, the R2 dose was certainly lower than doses achieving concentrations >50 µM at which peak INa blockade is observed. 8 In support, Rajamani et al demonstrated that ranolazine at 30 µM significantly slowed the recovery from inactivation of peak INa. 16 Again, these findings cannot entirely exclude that ranolazine, even at lower concentrations, might affect local conduction in the infarcted zone. Accordingly, Szél et al showed that even 20 µM ranolazine caused a dose-dependent significant decrease of Vmax in normal canine Purkinje fibers, papillary muscle, and M-cells. 10 In this sense, the effect of ranolazine on ventricular conduction would be expected to be enhanced in partially depolarized ischemic myocytes consequently to a higher number of Na+ channels being in the open state at these conditions.
Net membrane current densities for INa, INaL, and IKr were found to differ in normal and ischemically damaged myocytes depending on the time elapsing after coronary occlusion. This may obviously impact the ability of ranolazine to change conduction, repolarization, and refractoriness beyond a dose-dependent effect. In line, Jiang et al showed that delayed rectifier currents are decreased in the epicardial border zone overlying a 5 day-old infarct. 17 The authors demonstrated that in the infarct zone myocytes with the rapid (IKr) and slow (IKs) component of IK have reduced current densities compared to normal/healthy myocytes. However, if a reentrant pathway encompasses the noninfarcted myocardium, then the wavelength of the reentrant impulse might be prolonged by IKr inhibitors, thereby slowing the VT rate. In support, findings by Yao et al 18 in rats showed that 3 days after an infarct, the noninfarcted RV myocytes exhibited no reduction in IKr density.
Reduced dispersion of ventricular repolarization and refractoriness could also be accounted for the antiarrhythmic effects of ranolazine in the late post infarction period, most likely due to its transmurally different INaL/IKr inhibitory action. Furthermore, it has been suggested that in surviving cells in the 5-day infarcted heart, cellular inexcitability can outlast the repolarization phase of the action potential, 19 thus prolonging postrepolarization refractoriness. Unfortunately, none of the abovementioned mechanisms was able to prevent/delay spontaneous or induced VAs several days after MI. In our rabbit model, VERP was found to be shorter or very close to the MAPD90 values so that postrepolarization refractoriness was not observed neither in the control condition nor after ranolazine.
Although not systemically studied, in some experiments, triangulation-like MAP alteration has been observed only after R1. At the 30% repolarization level, R1 caused an abbreviation of MAP relative to control, possibly due to predominant INaL inhibition. However, a typical for triangulation phase 3 deceleration was not observed as by other drugs, for example, certain antibiotics or IKr blockers by reduced repolarization reserve. On the other hand, ranolazine at therapeutic concentrations seems to be a relative weak IKr blocker in ventricles producing only moderate prolongation of APD90.
The relationship of subacute-MI VAs and conduction inhomogeneity has been examined by Dillon et al who demonstrated that in the epicardial border zone of the 5-day infarcted heart, reentrant arrhythmias occur in fibers where the impulse propagation shows accentuated anisotropic properties. 20 Whether ranolazine affects this conduction anisotropy in surviving myocytes within the infarcted area or the infarct border zone, remains elusive. Further research is needed to clarify a dose-dependent effect of ranolazine on the conduction of the ischemic myocardium, including the gap junctional channel contribution, 21 in order to better understand its mechanism of action in the post-MI patients.
Limitations
A limitation of this study is that it did not examine the effect of R1 or R2 on action potential properties in the ischemic border zone of infarction. The fibrotic degeneration of the epicardial surface restricts the MAP recordings in that area in the beating heart model. The absence of assessments in plasma concentrations is an another limitation, however, this was partially counterbalanced by comparing similar dosing regimens of ranolazine in the same experimental species performed by previous investigators that determined the corresponding drug concentrations in blood. Finally, the induction of VAs using programmed stimulation is obviously not identical to the clinical states, where these arrhythmias initiate spontaneously; however, their course often resembles patterns of inducible VAs in experimental studies.
Conclusion
Ranolazine appears to acquire a dose-dependent antiarrhythmic effect on pacing-induced reentrant VAs during the late phase of MI in anesthetized rabbits. The higher dose of the drug only exerted substantial antiarrhythmic effects, whereas the lower dose regimen had no beneficial effects. In fact, there was even a tendency for an exacerbation of these arrhythmias with the lower ranolazine dose. A potential mechanism that might explain the ability of R2 to suppress reproducibility of VAs and/or to prolong sVT-CL is a pronounced increase of ventricular refractoriness possibly due to IKr blockade.
Although more investigation is still required to elucidate the electrophysiological mechanisms of ranolazine within the infarcted zone, the present findings suggest a dose-dependent antiarrhythmic effect of the drug after subacute MI in rabbits.
Footnotes
Acknowledgment
The authors are very indebted to I Makantasis for his technical assistance, and Dr Vet Z Arsenopoulou for inspecting the health status of the experimental animals.
Author Contributions
Aidonidis I. contributed to conception and design, acquisition, analysis, or interpretation, drafted the manuscript, gave the final approval, and agrees to be accountable for all aspects of work ensuring integrity and accurarcy; Moschovidis V. contributed to all experimental interventions, acquisition, analysis, and interpretation, and agrees to be accountable for all aspects of work ensuring integrity and accuracy; Simopoulos V. contributed to surgical procedure, and agrees to be accountable for all aspects of work ensuring integrity and accuracy; Stavela S. contributed to surgical procedures and anesthesia, analysis, and agrees to be accountable for all aspects of work ensuring integrity and accuracy; Dipla K. critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy; Hatziefthimiou A. contributed to acquisition, analysis, and interpretation, and agrees to be accountable for all aspects of work ensuring integrity and accuracy; Stamatiou R. contributed to acquisition, analysis, and interpretation, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship,and/or publication of this article.