
Abstract
The mechanisms for cardiac injury after hemorrhagic shock (HS) are unresolved. We hypothesize that remote organ damage can be caused by uncontrolled pancreatic proteolytic activity, as enteral protease inhibition improves outcomes in experimental HS. Uncontrolled proteolysis in the heart may disrupt cardiac metabolism and adrenergic control with subsequent deleterious outcomes. To test this hypothesis, the heart rate–pressure product (RPP) as an index of myocardial oxygen consumption and the levels of fatty acid transporter proteins CD36 and FATP6 as surrogates for metabolic activity in the heart were measured in rats subjected to experimental HS (n = 6/group) with and without the enteral protease inhibitor tranexamic acid (TXA). Plasma troponin I and heart fatty acid-binding protein (HFABP) concentrations were measured as indices of myocardial damage. Expression of the adrenergic receptors β1, α1D, and β2 was also measured in the heart to determine the possible effects of shock with and without enteral TXA on the adrenergic control of heart function. Hemorrhagic shock was induced by reduction in mean arterial blood pressure to 35 mm Hg for 2 hours before reperfusion of shed blood. The RPP was maintained in shocked animals treated enterally with TXA but not in those subjected to HS alone; this group also demonstrated decreased HFABP and plasma troponin I levels. Serine protease (trypsin, chymotrypsin, and elastase) and matrix metalloproteinase (MMP)-2 and MMP-9 activity was elevated in cardiac tissue and plasma after HS and abrogated by enteral TXA. Levels of CD36, FATP6, β1, α1D, and β2 were also increased after HS in cardiac tissue, and the increases were mitigated by TXA treatment. These results suggest that increased proteolytic activity may contribute to cardiac injury after HS. Enteral TXA prevents these changes, indicating a potential therapeutic option in the management of shock with resultant cardiac injury.
Introduction
Hemorrhagic shock (HS) is a leading cause of patient mortality and morbidity, 1 and even after resuscitation and hemodynamic stabilization, end-organ hypoperfusion often compromises outcomes of therapy. The mechanisms for devolution into multiple organ failure (MOF) after hemorrhagic shock are not completely understood; however, preservation of organ function by restoration and maintenance of cardiac performance and tissue perfusion is of paramount importance. Previous studies have demonstrated that cardiac performance is depressed in HS, 2 -4 but the mechanisms responsible for this are not completely understood. Cardiac function in vivo is modulated by several factors, including autonomic innervation and the chronotropic and inotropic responses to circulating mediators, while metabolism depends mainly on uptake of fatty acids. 5 Many of these processes are facilitated by cell surface transmembrane receptors such as the adrenergic receptors (e.g., β1, α1D, and β2) and fatty acid transporters including CD36 and FATP6. Despite the importance of these receptors in the maintenance of cardiovascular performance, variations in the density of hemodynamically and metabolically important transmembrane receptors have been incompletely studied in HS. 6 -8
Previous studies in a rat model of HS have demonstrated that proteolytic activity may be of importance in the evolution of the disease process. 9 -11 The origin of these proteases has been traced to the small intestine, where ischemia to the intestinal mucus membrane in experimental hypovolemic shock increases mucosal permeability and allows the escape of pancreatic enzymes from the gut lumen into the systemic circulation. 12,13 The effector enzymes in the circulation may include gut-derived pancreatic enzymes and/or matrix metalloproteinases (MMPs) activated by digestive proteases and other processes. Treatment of animals subjected to experimental shock with intralumenally administered protease inhibitors such as tranexamic acid (TXA) decreases protease activity in plasma and injury to principal organs and improves survival. 14 -16 We hypothesized that pathologic intravascular protease activation contributes to cardiac injury and that treatment with enteral TXA prevents this injury and improves outcomes. 17 To test the hypothesis, we used a rat model of HS with and without treatment with enteral TXA to investigate the rate pressure product (RPP) as an index of myocardial oxygen consumption in response to protease changes in the heart and plasma and measured the resultant changes in the expression of several cardiac transmembrane receptors known for their role in the regulation of cardiac and metabolic function.
Methods and Materials
Experimental Protocol
The animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Diego, and conforms to the Guide for the Care and Use of Laboratory Animals, 8th edition, by the National Institutes of Health (2011).
Twenty-four nonfasted male Wistar rats (300-450 g; Harlan Laboratories, Inc, Indianapolis, Indiana) were randomly assigned to control (no shock; n = 6), HS (n = 12, 6 with laparotomy and enteral control fluid injection and 6 without laparotomy or enteral fluid injection), or HS with enteral TXA treatment (HS+TXA; n = 6) groups. The animals were housed in the vivarium of the Department of Bioengineering at the University of California–San Diego. The temperature (20°C-22°C) and humidity (70%-75%) in the vivarium are controlled and continuously monitored, and the light–dark cycles (12 hours light / 12 hours dark) maintained according to standardized procedures for animal housing. Each cage houses at most 2 rats. Food and water are continuously available without fasting before the experiments. The health of the animals is visually verified before the experiments.
All rats were given general anesthesia (xylazine, 4 mg/kg; ketamine 75 mg/kg intramuscularly) with supplements administered as needed (xylazine, 4 mg/kg; ketamine 7.5 mg/kg intravenous). The right femoral vein and artery were cannulated for blood withdrawal and supplemental anesthesia and continuous monitoring of arterial pressure, respectively. Body temperature was maintained at 37°C via water-heated support and heat blanket.
After induction of anesthesia and vascular line placement, the animals were hemodynamically stabilized and heparinized (1U/mL estimated total blood volume as 6% of body weight) to prevent clotting of vascular lines. Hemorrhage was induced by blood withdrawal via the femoral vein (0.5 mL/min) to a target mean pressure of 35 mm Hg. Mean arterial blood pressure (MABP) was maintained between 30 and 40 mm Hg for 2 hours, after which time the withdrawn blood was warmed to 37°C, and animals were reperfused (0.5 mL/min) and monitored for an additional 2 hours. Control animals were treated in analogous fashion without the removal of blood.
A laparotomy (trauma) was performed 1 hour into the hypovolemic period on the HS (n = 6) and HS+TXA (n = 6) animals. In the HS+TXA group, 127 mmol/L TXA (Cyclokapron, Pfizer) was administered into the lumen of the small intestine via sequential injections longitudinally along the length of the small bowel. 0.14 g GoLytely (Braintree Laboratories Inc, Braintree, MA, USA)/mLin 0.9% normal saline (NS) was used as carrier solution. A total fluid volume of 15 mL was injected into the lumen of the small intestine. An additional 2 mL of TXA was injected into the cecum. The dosage of the TXA was based on previously determined effective concentrations. 14 The HS animals (n = 6) received the identical treatment with enteral GoLytely in NS only.
A second group of animals undergoing HS (n = 6) was instrumented as above but without laparotomy or subsequent fluid injection into the bowel to control for possible confounding effects of laparotomy and fluid injection into the bowel. All ex vivo analysis was conducted between control, this second HS group, and HS+TXA groups.
Following a 2-hour observation period after the return of shed blood (Reperfusion) or after the analogous time period in the control group, the animals were euthanized with B-Euthanasia (120 mg/kg). Death was confirmed by verification of cardiac arrest after bilateral thoracotomy.
Plasma Collection and Tissue Harvest
Immediately prior to euthanasia 1 mL venous blood was collected and centrifuged at 1000 g for 10 minutes. Plasma was collected and stored at −80°C for later use in gelatin zymography, Western blot analysis, and the HFABP and troponin I assays. The heart was harvested following thoracotomy and the ventricles sectioned in 3 transversal sections, with the apex stored at −80°C for later use and 2 other sections used for immunohistochemical (IHC) analysis or homogenized in lysis buffer for electrophoresis. The cardiac tissue was rinsed after collection to minimize the amount of residual blood in the tissue and the presence of blood-borne enzymes. Homogenate was divided into 2 aliquots. The first aliquot was stored for gelatin zymography, while the second one was treated with the addition of a protease inhibitor cocktail (cOmplete Protease Inhibitor Cocktail; Roche Applied Sciences, Mannheim, Germany) for Western blot analysis. Heart homogenates were stored at −80°C for later use.
Plasma and Heart Homogenate Gelatin Zymography
To determine proteolytic activity in the plasma and heart samples for each animal (1.6 µL for plasma and 5 µL at 4 µg protein/µL for heart), cardiac homogenates and plasma were separated by gel electrophoresis in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels containing 80 µg/mL gelatin. Gels were renatured by four 15-minute washes with 2.5% Triton X-100 and incubated overnight at 37°C in developing buffer (0.05 mol/L Tris base, 0.2 mol/L NaCl, 4 µmol/L ZnCl2, and 5 mmol/L CaCl2·2H2O). After incubation, gels were fixed and stained (50% methanol, 10% acetic acid, 40% water, and 0.25% Coomassie blue solution) for 1 hour before destaining in buffer (10% methanol, 10% acetic acid, and 80% water). The molecular weights of the proteases were estimated by use of a standard protein ladder (Invitrogen, Carslbad, CA, USA). Gels were digitized and bands were digitally analyzed by densitometry (NIH Image J software version 1.44P; http://imagej.nih.gov/ij/).
Western Blot Detection of Plasma and Heart Proteins
Plasma and heart homogenate samples from each animal (3 µL/sample for plasma and 20 µL/sample at 4 µg/µL for heart) were separated by SDS-PAGE. For Western blotting, proteins were transferred to a 0.45-μm pore size nitrocellulose membrane (Bio-Rad Laboratories, Inc, Hercules, California; #162-0097). Following blockade with 5% nonfat dry milk in Tris-buffered saline Tween 20 (TBST), primary polyclonal antibodies against MMP-2 (H-76, 1:100, sc-10736; Santa Cruz Biotechnology, Santa Cruz, California), MMP-9 (H-129, 1:100, sc-10737; Santa Cruz Biotechnology), trypsin (D-1, 1:100, sc-137077, Santa Cruz Biotechnology), chymotrypsin (T-20, 1:100, sc-69255; Santa Cruz Biotechnology), elastase (M-18, 1:100, sc-9521; Santa Cruz Biotechnology), and heart fatty-acid binding protein (HFABP; 1:1000, ab45966; Abcam, Cambridge, United Kingdom) were applied to the plasma and heart homogenate blots. Additionally, primary antibodies against fatty acid transport protein-6 (FATP6; 1:1000; ab167099; Abcam), CD36 (1:1000; ab80978; Abcam), β1 adrenergic receptor (1:1000; ab3442; Abcam), α1D adrenergic receptor (H-142, 1:300, sc-10721; Santa Cruz Biotechnology), and β2 adrenergic receptor (H-20, 1:300, sc-569; Santa Cruz Biotechnology) were applied to heart homogenate blots. β-Actin (C4, 1:100, sc-47778, Santa Cruz Biotechnology) served as loading control. Blots were restripped and reprobed for target proteins (Restore Western Blot Stripping Buffer; Thermo Scientific, Rockford, IL, USA; Life Technologies). Corresponding secondary antibodies were applied at 1:5000 dilution. All antibodies were diluted in 5% non fat milk in TBST. Supersignal West Pico Chemiluminescent substrate (ThermoFisher Scientific, Waltham, Massachusetts; #34096) was used for imaging. The predicted molecular weight bands were used to confirm the location of the protein on the blot according to antibody manufacturer specifications. The molecular weights of the proteases were estimated by use of an electrophoresis marker (C1992, Sigma Aldrich, St Louis, Missouri, USA). Gels were digitized and bands were analyzed by digital densitometry (ImageJ).
Troponin I Assay
Plasma samples (100 µL) from control, HS, and HS+TXA groups (n = 6) were used to measure circulating cardiac troponin I levels using a standard enzyme-linked immunosorbent assay (MBS727624, Mybiosource, San Diego, California). Concentrations were calculated from absorbance measurements and a standard curve according to the kit protocol.
Immunohistochemistry
Fresh cardiac tissue was fixed (10% formalin) for sectioning after the collection of 3 transversal ventricular regions following heart harvesting as explained earlier, and 30-μm thick sections were made for IHC analysis. The sections were labeled with the same primary antibody used for Western blot. Five antibodies were tested: against the FATP6 receptor, the CD36 receptor, the β1 adrenergic receptor, the α1D adrenergic receptor, and the β2 adrenergic receptor. Dilution for all antibodies was 1%. The sections were then stained with ImmPACT NovaRED Peroxidase substrate (Vector Laboratories, Burlingame, California) and incubated for 30 seconds. Images of label density were taken at 20× objective magnification and digitally recorded for measurement of light absorption (ImageJ).
A standard hematoxylin and eosin stain was also performed to characterize inflammatory cell accumulation and the presence of micro-hemorrhages in the heart tissue.
Statistical Analysis
Two-sided Student t test was used for comparison between 2 continuous variables, while 1-way analysis of variance was used where appropriate to evaluate the differences between groups with post hoc Tukey correction. P < .05 was considered significant. All statistical analyses were performed using Graphpad (Graphpad Software Inc, La Jolla, California).
Results
Systemic Hemodynamics
The RPP, defined as MABP × HR, was not significantly different between HS and HS+TXA groups initially nor during the hypotensive period but increased significantly in HS+TXA animals upon reperfusion of shed blood (Figure 1A). MABP is increased and restored in the HS+TXA group following resuscitation (Figure 1B). Heart rate remains elevated compared to baseline after reperfusion in both shock groups (Figure 1C). There were no significant differences in RPP between HS groups that received and did not receive laparotomy and enteral fluid (Supplemental Figure 1).
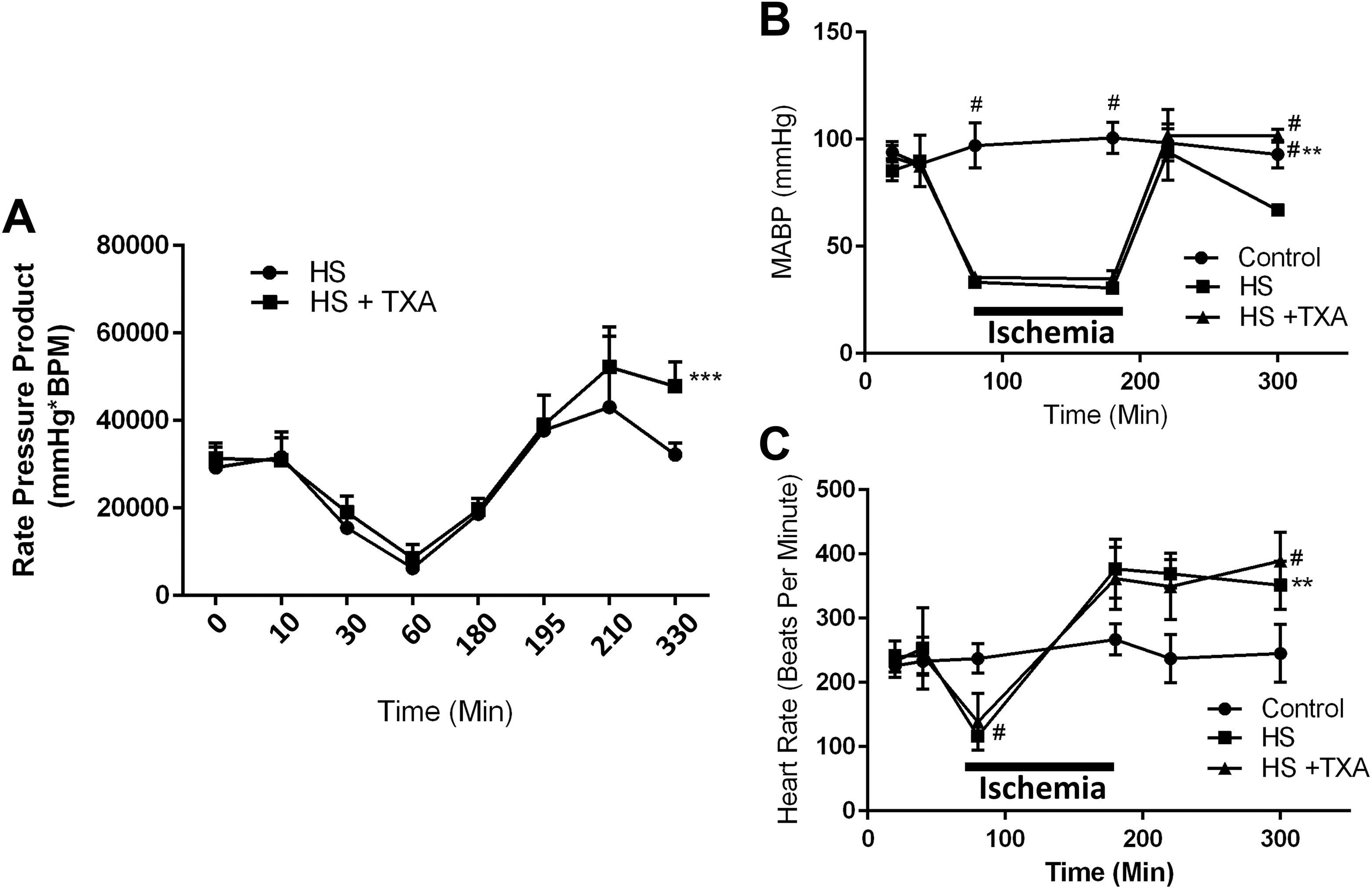
(A) Rate pressure product (RPP = MABP × HR). (B) Mean arterial blood pressure (MABP) and (C) heart rate (HR) in experimental hemorrhagic shock (HS). The MABP was decreased to a mean of 35 mm Hg over several minutes by the serial withdrawal of small aliquots of blood (0.5 mL/min). Reperfusion was carried out in analogous fashion by slow infusion of shed blood (0.5 mL/min). ***P < 0.001 HS vs HS+ tranexamic acid (TXA) for RPP; ** P < 0.01 control vs HS+TXA MABP, and #P < 0.0001 HS vs HS+TXA and control MABP; comparisons for heart rate: **P < 0.01 control vs HS, #P < 0.0001 control vs HS, HS+TXA during ischemia period, and control vs HS+TXA at reperfusion. (n = 6 rats/group).
Plasma Protease Levels
Serine protease activity in plasma as measured by zymography was significantly increased in HS compared to control and decreased by enteral TXA (Figure 2A). Enteral TXA administration (HS+TXA) also reduced plasma concentrations of trypsin, chymotrypsin, and elastase (Figure 2B-D). Plasma MMP-2 activity, as measured by zymography, was also significantly increased in HS compared to control and abrogated by enteral TXA treatment (Figure 3) with a similar trend in plasma protein content between the groups. Plasma MMP-9 showed a trend similar to MMP-2 but was not significantly different between control and the HS groups.

Plasma serine protease activity and band density by gelatin zymography and Western blot. Average control band density used a reference. (A) Plasma serine protease activity, *P < 0.05 hemorrhagic shock (HS) vs HS+ tranexamic acid (TXA), **P < 0.01 HS vs control; (B) Trypsin band densities, **P < 0.01, HS vs control, HS+TXA; (C) chymotrypsin band densities, *P < 0.05, HS vs HS+TXA; and (D) elastase band densities, *P < 0.05, control vs. HS, **P< 0.01 HS vs HS+TXA.

Plasma matrix metalloproteinase (MMP)-2 and MMP-9 activity and band density by gelatin zymography and Western blot. All values normalized to control average. (A) Plasma MMP-2 activity, *P < 0.05 hemorrhagic shock (HS) vs HS+ tranexamic acid (TXA), **P < 0.01 control vs HS; (B) MMP-2 band densities. (C) Plasma MMP-9 activity, dark bands correspond to non-active protein; (D) MMP-9 band densities. (n = 6 rats/group).
Cardiac Protease Levels
Zymography of cardiac tissue displayed significantly increased serine protease concentration and activity after HS (Figure 4A and B). This activity was completely inhibited by enteral TXA treatment. Protein content of serine proteases in cardiac tissue mirrored results from plasma, with increases in trypsin measured after hemorrhagic shock but not chymotrypsin. Endogenous activation of both MMP-2 and MMP-9 was also significantly increased in cardiac tissue in HS; enteral treatment with TXA significantly reduced this activity to control levels without significant changes in total protein content (Figure 4C and D).

Cardiac serine protease activity and band density by gelatin zymography and Western blot. (A) Serine protease activity bands. The average of the control band densities was used as a reference, **P < 0.01 control vs hemorrhagic shock (HS), *** P < 0.001 HS vs HS+ tranexamic acid (TXA). Band density measurements for (B) trypsin, * P < 0.05 control vs HS, chymotrypsin, and elastase. (C) Cardiac matrix metalloproteinase (MMP)-2 and 9 activity and band density by gelatin zymography and Western blot. All values normalized compared to control average. *P < 0.05 HS vs control and HS+TXA and cardiac MMP-2 band densities; (D) Cardiac MMP-9 activity, ** P < 0.01 control vs HS, *** P < 0.001 HS vs HS+TXA; and cardiac MMP-9 densities. (n = 6 rats/group).
Circulating and Endogenous HFABP and Troponin I Levels
The acute heart failure marker HFABP was significantly increased in HS in cardiac tissue and plasma as detected by immunoblotting (Figure 5A and B). Enteral treatment with TXA mitigated this increase. Likewise, plasma levels of Troponin I, another indicator of cardiac damage, significantly increased in HS and decreased to near baseline by treatment with TXA (Figure 5C). Microscopic examination confirms post-shock changes with increased number of inflammatory cells and microhemorrhages seen in cardiac tissue after HS compared to TXA-treated animals (Supplemental Figure 2).

Cardiac and plasma heart fatty acid-binding protein (HFABP) band intensities and plasma troponin I concentrations. Average control band density used as reference. (A) Cardiac HFABP band densities, * P < 0.01 control vs hemorrhagic shock (HS) and (B) plasma HFABP band densities. (C) Plasma Troponin I levels, *** P < 0.0001 control vs. HS, ** P < 0.001 HS vs HS+ tranexamic acid (TXA). (n = 6 rats/group).
Fatty Acid Transport Protein and Cardiac Adrenergic Receptor Levels in the Heart
The densities of both fatty acid transporters FATP6 and CD36 increased significantly in HS, and this increase was prevented by enteral treatment with TXA as determined by both Western blot and IHC (Figure 6).
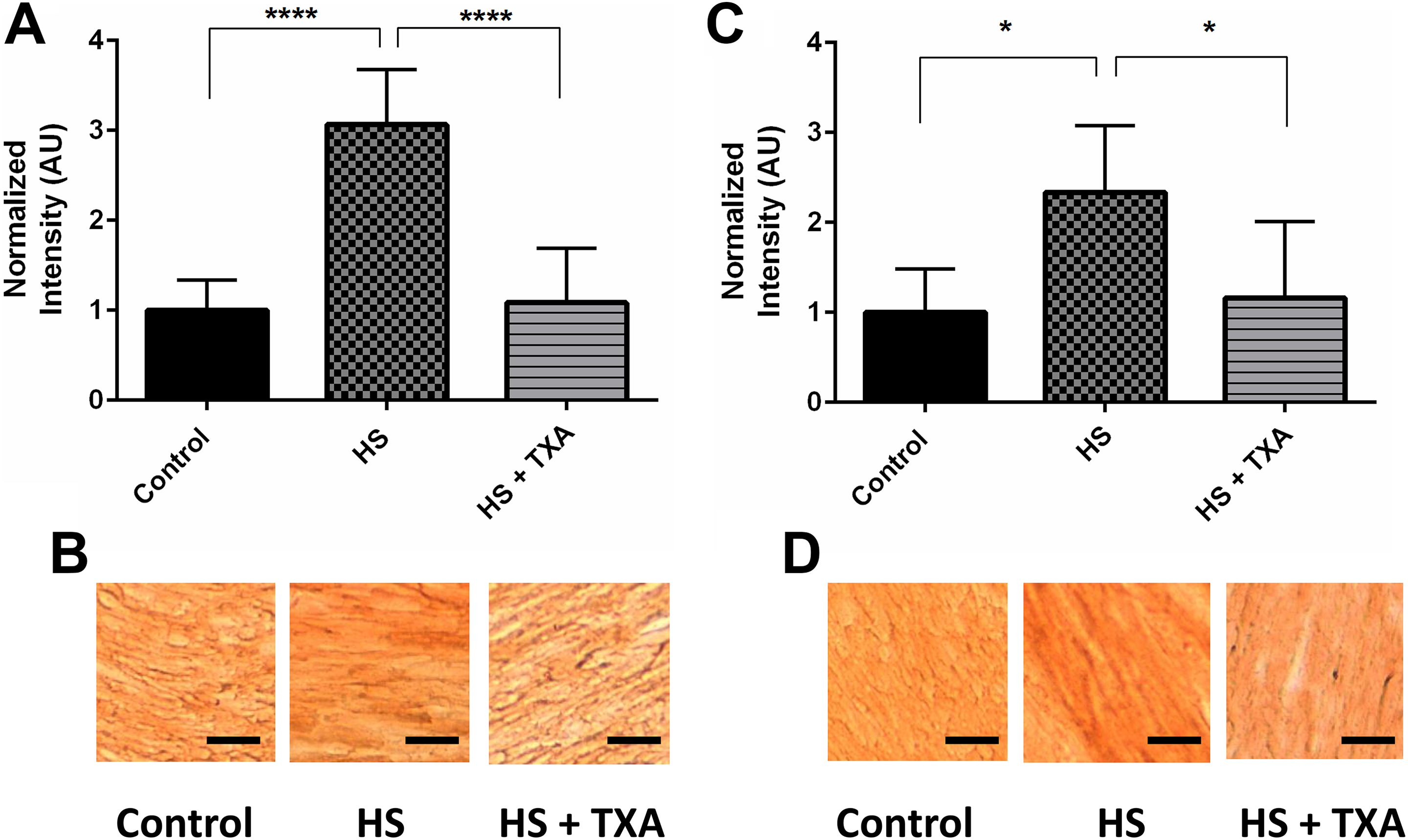
Metabolic receptors CD36 and FATP6 densities by Western blot and immunohistochemical staining for CD36 and FATP6. Average control band density used as reference. (A) CD36 receptor band densities, ****P < 0.0001 hemorrhagic shock (HS) vs control, HS+ tranexamic acid (TXA). (B) CD36 staining of control, HS, and HS+TXA myocardium. (C) FATP6 receptor band densities, * P < 0.05 HS vs control, HS+TXA. (D) FATP6 staining of control, HS, and HS+TXA myocardium. Black bar = 100 µm. (n = 6 rats/group).
Levels of cardiac β1 and α1D adrenergic receptors were significantly increased in HS. TXA reduced adrenergic receptor density values to near baseline with similar trends as measured by both Western blot and IHC (Figure 7).

Cardiac β1, α1D, and β2 adrenergic receptor band densities by Western blot and immunohistochemical staining for β1, α1D, and β2 adrenergic receptors. Average control band density used a reference. (A) β1 adrenergic receptor band densities, * P < 0.05 control vs hemorrhagic shock (HS) and *P < 0.05 control vs HS+ tranexamic acid (TXA); (B) β1 staining of control, HS, and HS+TXA myocardium; (C) α1D adrenergic receptor band densities, *P < 0.05 control vs HS and *P < 0.05 HS vs HS+TXA; (D) α1D staining of control, HS, and HS+TXA myocardium; (E) β2 adrenergic receptor band densities; (F) β2 staining of control, HS, and HS+TXA myocardium. Black bar = 100 µm. (n = 6 rats/group).
Discussion
Cardiac dysfunction has long been recognized as a serious complication of HS. 18 -22 The mechanisms of hemorrhage-induced cardiac injury and failure are still poorly understood but may be caused by gut-derived mediators transported predominantly by the lymphatics. 17,23,24 Enteral TXA has previously been shown to decrease morbidity and mortality in experimental HS by preventing proteolytic degradation of the small bowel mucosa and increases in gut permeability to inflammatory mediators and digestive proteases. 14 We hypothesized that enteral TXA prevents cellular injury affecting the heart and other organs in shock, and we designed this study to investigate the potential involvement of proteases in modulating metabolic-related cardiac changes (RPP, CD36, and FATP6), cell injury (Troponin I and HFABP), and the expression of surface receptors that play a role in the regulation of cardiac function.
Tranexamic acid was used as the enteral protease inhibitor for these studies for several reasons. Although not a protease inhibitor in the classic sense, TXA partially inhibits trypsin (in addition to its well-known actions against plasminogen and in higher concentrations plasmin). Unlike many serine protease inhibitors, TXA has a high therapeutic index and is well known in the trauma world. Importantly for potential clinical use, it is Food and Drug Administration approved.
We demonstrate here that enteral treatment with TXA in HS improves RPP by maintaining hemodynamic stability and prevents cardiac injury as measured by Troponin I and HFABP changes. CD36 and FATP6 levels were also reduced to near baseline in HS after enteral treatment with TXA, suggesting preservation of metabolic cardiac homeostasis. Taken together, these results suggest that enteral TXA treatment mitigates cardiac injury and the requirement for subsequent pathophysiologic compensatory mechanisms in the heart that are otherwise seen in HS.
Inhibition of bowel-derived proteases may be a possible mechanism for this protection, as experimental HS results in increases in systemic gut-derived proteases and circulating MMPs 25 in plasma and cardiac tissue which are decreased by enteral TXA, although the mechanisms behind this protection are not entirely clear.
Data from our laboratory indicate that levels and functionality of the α1D receptor are decreased in the vasculature in hemorrhagic shock. 26 Therefore, we anticipated a similar decrease in cardiac concentrations of this receptor as well as the β1 and β2 adrenergic receptors and additional catecholamine receptors examined for their role in cardiac function. 27 However, these receptor concentrations increased in HS in the heart, mirroring similar increases seen in the other transmembrane receptors studied.
Treatment with enteral TXA decreased the concentrations of the fatty acid transporters and the adrenergic receptors, suggesting a compensatory response to hemorrhagic shock that is blunted by TXA treatment. 8,22,28 To correlate cardiovascular performance with cardiac injury, we measured HFABP and Troponin I, 2 specific markers for cardiac injury. 29,30 Increases in both tissue and plasma HFABP and in plasma Troponin I in HS confirm cardiac tissue damage in our model of HS, which was mitigated by enteral TXA treatment. 31
There are several limitations to our study. First, TXA is a well-known antifibrinolytic that is used to control bleeding and has proven beneficial in reducing mortality in HS. 32 In our enteral approach, we hypothesize that TXA is acting primarily in the gut as a (partial)-serine protease inhibitor rather than as an antifibrinolytic in the systemic circulation. However, we cannot discount that enteral TXA has concomitant salubrious effects in the systemic circulation that may or may not involve the coagulation protease cascade.
Further, although we confirmed the association between gut-derived proteolytic activity in the heart and cardiac injury and decreased hemodynamic stability in HS, which are prevented by enteral protease inhibition, we have not established whether increases in proteolytic activity are causal to cardiac dysfunction. Our results implicate a possible improvement in cardiac performance in the TXA-treated groups, although this interpretation requires confirmation by direct measurements of heart contractility. However, MABP was maintained in the TXA-treated group after reperfusion, hinting at improved cardiovascular performance from the treatment. Still, it remains to be demonstrated whether improved hemodynamics are due to improved cardiac performance, maintained vascular integrity, or, most likely, a combination of both.
Our results may not be generalizable to cardiac receptors in general, as different receptors may respond in a temporal or tissue-specific manner. There are very few studies, however, addressing HS and receptor expression in the heart. Toll-like receptor 4 in the failing myocardium is often overexpressed, 33 while β1 is downregulated in experimental HS at 6 hours. 7 Further studies are necessary to determine the relative importance of these findings. Our study focused on acute changes in cardiac receptor density and function occurring in response to HS with and without enteral TXA; it is unclear whether the changes measured in our study are sustained or how they may modulate in time. Finally, although we demonstrate decreased cardiac injury after treatment with TXA and improved cardiovascular stability, we did not directly confirm changes in cardiac performance in this study.
Conclusion
In summary, serine protease and MMP activity in plasma and the heart, as well as important metabolic and hemodynamic transmembrane receptors, cardiac injury, and hemodynamic instability, are increased in experimental HS. These changes are prevented by enteral TXA treatment in accordance with the “autodigestion” hypothesis that intestinally-derived serine proteases become systemic in circulatory shock. 34 These results suggest that enteral protease inhibition, by prevention of systemic protease activation, protects against cardiac injury and aids in maintaining hemodynamic stability and improved outcomes in HS.
Supplemental Material
Supplemental_Figure1 - Enteral Tranexamic Acid Decreases Proteolytic Activity in the Heart in Acute Experimental Hemorrhagic Shock
Supplemental_Figure1 for Enteral Tranexamic Acid Decreases Proteolytic Activity in the Heart in Acute Experimental Hemorrhagic Shock by Federico Aletti, Marco Santamaria, Kevin Chin, Rafi Mazor and Erik B. Kistler in Journal of Cardiovascular Pharmacology and Therapeutics
Supplemental Material
Supplemental_Figure_2 - Enteral Tranexamic Acid Decreases Proteolytic Activity in the Heart in Acute Experimental Hemorrhagic Shock
Supplemental_Figure_2 for Enteral Tranexamic Acid Decreases Proteolytic Activity in the Heart in Acute Experimental Hemorrhagic Shock by Federico Aletti, Marco Santamaria, Kevin Chin, Rafi Mazor and Erik B. Kistler in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Authors’ Note
The contents do not represent the views of the U.S. Department of Defense, Department of Veterans Affairs or the United States Government.
Author Contribution
Federico Aletti, Kevin Chin, and Erik B. Kistler contributed to acquisition, analysis, or interpretation; contributed to conception or design; drafted the manuscript; critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring integrity and accuracy. Marco Santamaria contributed to acquisition, analysis, or interpretation; contributed to conception or design; critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring integrity and accuracy. Rafi Mazor contributed to conception or design; gave final approval; and agreed to be accountable for all aspects of work ensuring integrity and accuracy. Federico Aletti and Marco Santamaria contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by: the European Union “CelSys Shock” Marie Curie International Outgoing Fellowship PIOF-GA-2012-328796; the European Union “ShockOmics” grant #602706; the Department of Defense Award W81XWH-17-2-0047; the Career Development Award (CDA2) 1IK2BX001277-01A1 from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development; and the NIH grant GM085072-06.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.