
Abstract
P2Y12 receptor-blocking drugs given at reperfusion offer protection against myocardial infarction in animal models of transient coronary occlusion. Two recent reports concluded that ticagrelor was more cardioprotective than clopidogrel and attributed this to ticagrelor’s unique ability to raise tissue adenosine by blocking the equilibrative nucleoside transporter 1. Indeed, an adenosine receptor blocker attenuated ticagrelor’s protection. The related P2Y12 inhibitor cangrelor, which does not block the transporter, protects hearts only when platelets are in the perfusate, while adenosine is known to protect equally in situ blood-perfused and crystalloid-perfused isolated hearts. We, therefore, tested whether ticagrelor liberates a sufficient amount of adenosine to protect a Krebs buffer-perfused isolated rat heart subjected to 40 minutes of global ischemia followed by 2 hours of reperfusion. In untreated hearts, 77.6% ± 4.0% of the ventricle was infarcted as measured by triphenyltetrazolium staining. Ischemically preconditioned hearts had only 32.7% ± 3.6% infarction (P < .001 vs untreated), indicating that our model could be protected by preconditioning which is known to involve adenosine. Strikingly, hearts treated with 10 μmol/L ticagrelor in the buffer throughout the reperfusion period had 77.5% ± 2.4% infarction comparable to unprotected controls (P = NS vs untreated). These data strongly suggest that ticagrelor was unable to release sufficient adenosine from the crystalloid-perfused rat heart to protect it against infarction. Our previous studies have found no difference in the anti-infarct potency among clopidogrel, cangrelor, and ticagrelor in open-chest rats and rabbits, and surprisingly adenosine receptor antagonists block protection from all 3 drugs. We have no explanation why ticagrelor is more protective in the pig than clopidogrel but suspect a species or perhaps a treatment schedule difference that may or may not involve adenosine.
Today P2Y12 antagonists, which block binding of adenosine diphosphate (ADP) to platelet receptors, are standard of care in patients with acute myocardial infarction (AMI) whose arteries are stented during recanalization by primary percutaneous intervention. While the initial use of these antiaggregant drugs was based on their anticoagulant properties, emerging evidence has revealed pleiotropic effects that could be even more important than anticoagulation. These studies clearly demonstrate that P2Y12 antagonists clopidogrel, ticagrelor, and cangrelor are all potent postconditioning mimetics. When administered just a few minutes before reperfusion in acute animal models of myocardial ischemia/reperfusion (I/R), either ticagrelor 1 –3 or cangrelor, 2,4 both direct antagonists of the P2Y12 receptor, significantly diminish myocardial infarct size. Clopidogrel, however, is a prodrug and must be enzymatically converted to the active P2Y12 ligand before it can protect. When it is given just prior to reperfusion in animal models, it is not protective. 1 However, if sufficient time is allowed for conversion to its active form as evidenced by blockade of platelet reactivity, then clopidogrel does become cardioprotective. 4,5 Vilahur et al pretreated pigs with clopidogrel or ticagrelor several hours before the ischemic insult so that robust platelet inhibition occurred in both groups, and notably the anti-infarct effect of ticagrelor was greater than that of clopidogrel. 5 In our rabbit model, we saw equal protection from cangrelor or 2-day pretreatment with clopidogrel at doses that completely abolished ADP-induced platelet aggregation. 4 In contrast, Nanhwan et al 6 reported that treatment for 1 week with either ticagrelor or clopidogrel caused similar attenuation (but not complete abolition) of platelet aggregation in rats, but only ticagrelor was cardioprotective. Whether clopidogrel’s failure to limit infarct size was due to a species difference or tachyphylaxis from prolonged exposure or perhaps inadequate attenuation of aggregation is unknown.
In addition to its action as a P2Y12 antagonist, ticagrelor blocks the equilibrative nucleoside transporter (ENT) 1 that raises the tissue adenosine level. 7 Two groups have recently suggested that this noncanonical action of ticagrelor might explain the observation that ticagrelor is a more potent cardioprotectant than clopidogrel that has no effect on ENT 1. 1,5,8 Proof of this suggestion largely rests on the ability of a nonselective adenosine receptor antagonist to block ticagrelor’s infarct-sparing effect, but an adenosine receptor antagonist also blocked the protection from prolonged clopidogrel treatment. 4
The actual mechanism(s) whereby P2Y12 inhibition triggers conditioning’s protective signaling remains unknown, but it appears to involve platelets. Cangrelor will not protect either a blood-free, isolated heart 4 or the heart of a thrombocytopenic rat. 9 However, increasing adenosine concentration can trigger protection against infarction in the blood-free isolated heart. 10 To objectively evaluate the potential role of ticagrelor-mediated inhibition of ENT 1 in cardioprotection, ticagrelor’s effectiveness as a cardioprotectant was examined in a buffer-perfused isolated rat heart in which the drug’s putative effect on tissue adenosine could be separated from its action as a P2Y12 receptor antagonist because platelets are absent from the system.
Methods
All protocols were approved by the Institutional Animal Care and Use Committee of the University of South Alabama College of Medicine and conformed to published guidelines. 11
Surgical Preparation
Male Sprague-Dawley retired breeder rats weighing 400 to 600 g were anesthetized with intraperitoneal pentobarbital (100 mg/kg) and then ventilated with a positive pressure respirator and 100% oxygen. Through a left thoracotomy in the fourth intercostal space, the heart was quickly removed by cutting the great vessels and mounted on a Langendorff apparatus. The aorta was retroperfused under constant pressure with oxygenated, cell-free Krebs buffer. A fluid-filled balloon was placed in the left ventricular lumen, and the ventricular pressure was recorded. Global ischemia was created by arresting aortic perfusion for 40 minutes. The heart was kept at constant temperature by immersing in a water-jacketed chamber filled with buffer maintained at 38°C. The heart was subsequently reperfused for 2 hours. Immediately after cardiac extirpation, blood filling the thoracic cavity was collected in a heparinized syringe for platelet aggregometry as detailed subsequently.
Measurement of Infarct Size
In these hearts subjected to global ischemia, the entire ventricular mass is the risk zone. The heart was removed from the Langendorff apparatus, briefly frozen, and then sliced from apex to base into 1-mm-thick slices, which were incubated in warm triphenyltetrazolium chloride (TTC) solution to visualize the infarct (TTC-negative tissue). The slices were digitally photographed, and images were prepared for analysis. Borders of the ventricular slices and infarct regions were determined and areas calculated by Image-J software. The observer making these measurements was blinded to the treatment applied to each heart. Infarct size was expressed as a percentage of the risk zone which in this case was the entire left ventricle.
Platelet Aggregometry
Platelet aggregation was determined by measuring impedance with a whole blood aggregometer (Chrono-log Corp., Havertown, Pennsylvania). One-half milliliter of saline or buffer and 0.5 mL of heparinized blood were combined in a plastic cuvette and continuously stirred at 37°C. Aggregation was initiated by addition of 10 μL of 1 mmol/L ADP to produce a final concentration of 10 μmol/L. Area under each aggregation curve was measured, and areas were averaged. Aggregation curves were measured only in the groups treated with ticagrelor. For these experiments, platelet aggregation was determined with blood collected when the heart was removed, and the blood in the cuvette was diluted with either saline (control) or ticagrelor-containing buffer removed from the perfusion reservoir.
Protocols
Four groups of rats were studied. In the control group (n = 5), the perfusing buffer contained no additives. In the next 2 groups, ticagrelor was added to the buffer so that the final concentration was either 3 (n = 5) or 10 (n = 5) μmol/L. The hearts were perfused with standard buffer before cessation of aortic retroperfusion and initiation of global ischemia. After 40 minutes of ischemia, aortic retroperfusion was resumed for 2 hours with ticagrelor-supplemented buffer. In the fourth group (n = 5), hearts were ischemically preconditioned by arresting aortic perfusion for 5 minutes and then resuming flow for 5 minutes before a second cycle of ischemia/reperfusion. After 3 cycles, aortic retroperfusion was arrested for 40 minutes and then resumed for 2 hours as mentioned earlier.
Statistics
Infarct sizes were analyzed by 1-way analysis of variance. Post hoc testing was done with Student-Newman-Keuls test. Platelet aggregation curves were analyzed by determining area beneath the curves truncated at 5 minutes. Paired t test was used to compare average areas for control and ticagrelor treatment. A P value of <.05 was considered to be significant.
Results
Baseline heart rate and left ventricular developed pressure were not different in the 4 groups (data not shown). During global ischemia, the hearts stopped beating but cardiac contractions resumed upon reperfusion, and some developed pressure returned in all hearts. Platelet aggregation in whole blood was almost totally blocked by ticagrelor as a verification of its efficacy. Representative platelet aggregation curves are shown in Figure 1.
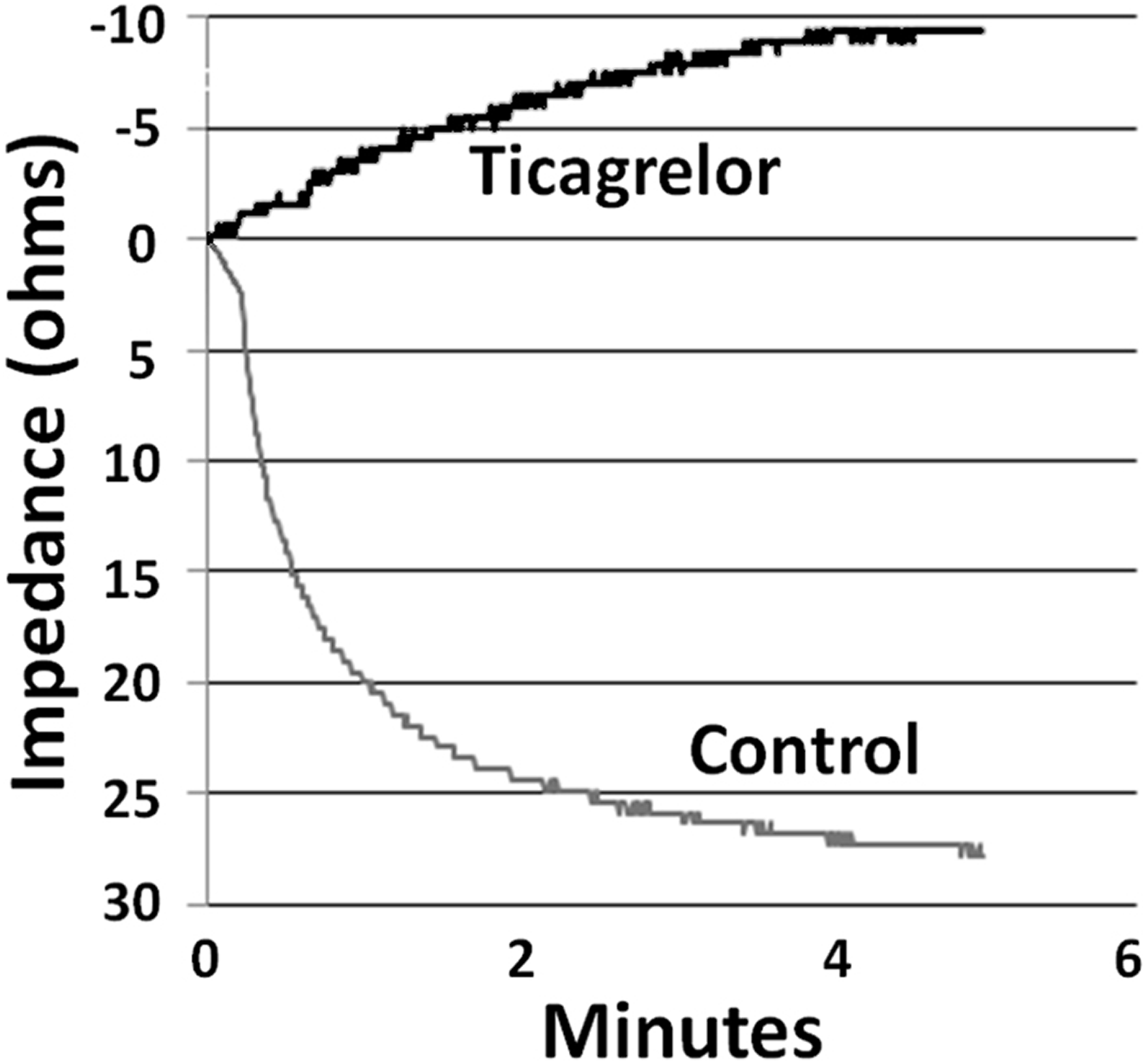
Platelet aggregation curves in a representative experiment. For the control curve, blood was mixed with Krebs buffer in the cuvette to which adenosine diphosphate (ADP) was added to initiate aggregation. For the ticagrelor curve, blood was mixed with buffer containing 10 μmol/L ticagrelor. The control aggregation curve shows the result of progressive aggregation over a 5-minute interval. The ticagrelor curve shows that aggregation was completely blocked and demonstrates only drift.
Infarct size in control, untreated hearts averaged 77.6% ± 4.0% of the risk zone after 40 minutes of global ischemia and 2 hours of reperfusion (Figure 2). Neither 3 nor 10 μmol/L ticagrelor, which demonstrated potent inhibition of platelet aggregation activity in whole blood, affected the amount of infarction (79.1% ± 2.7% and 77.5% ± 2.4%, respectively). Isolated hearts did appear to respond to adenosine because ischemic preconditioning as a positive control significantly decreased infarct size to 32.7% ± 3.6% of myocardium at risk (P < .001). Figure 3 shows 3 typical hearts sectioned into 1-mm-thick TTC-stained slices. Thus, our experiments did not detect any protection against infarction from ticagrelor in the isolated rat heart, and hence failed to demonstrate adenosine-mediated cardioprotection from ticagrelor.

Percentage of the left ventricle infarcted after 40 minutes of global ischemia and 2 hours of reperfusion in Krebs-perfused isolated hearts when exposed to either ischemic preconditioning (IPC) or 1 of 2 doses of ticagrelor. Open symbols represent data from individual hearts, whereas closed symbols represent group averages ± standard error of mean (SEM). Only ischemic preconditioning offered any protection against infarction as compared to untreated hearts or those exposed to ticagrelor. *P < .001 vs other groups.
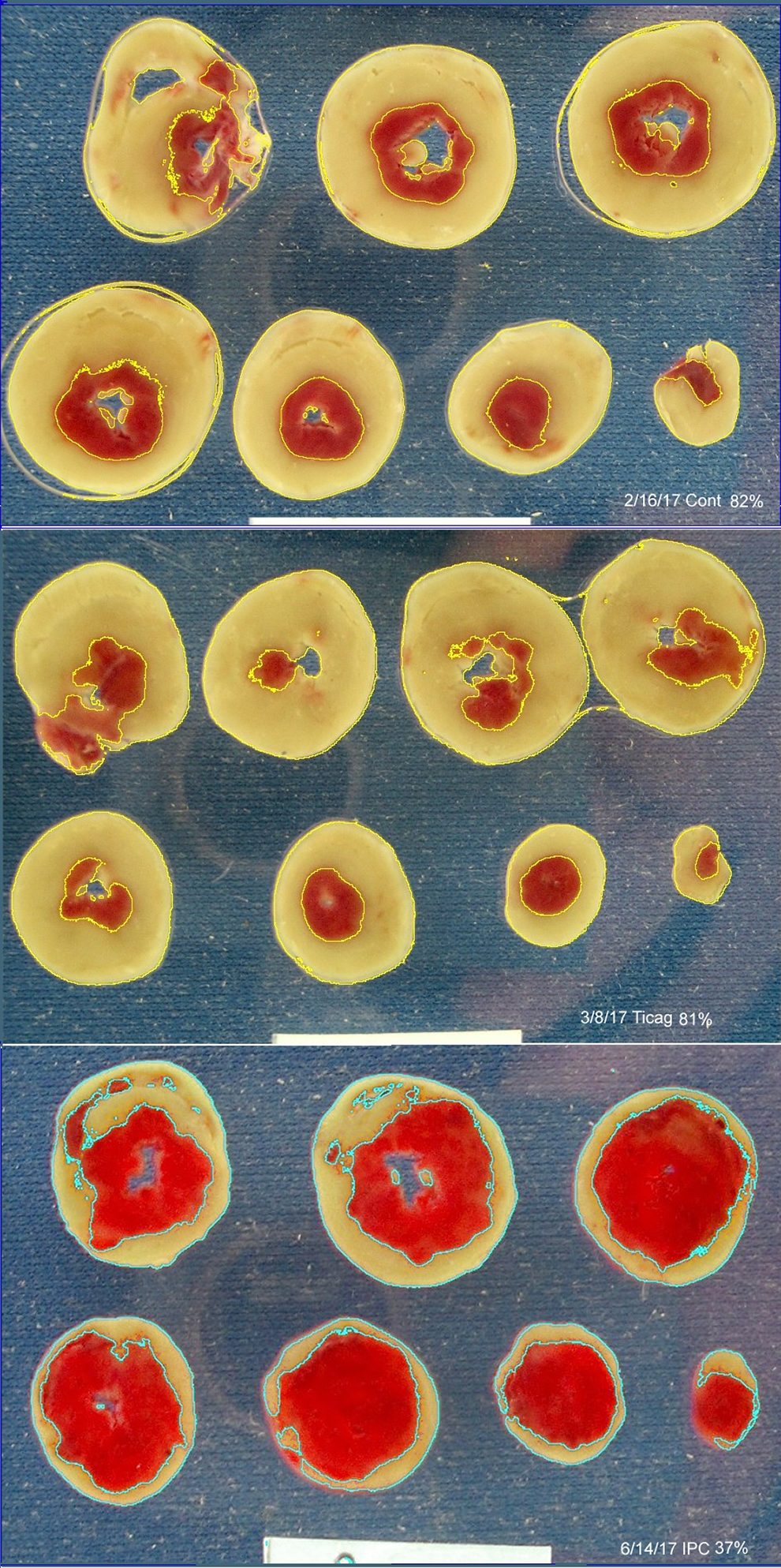
Composite of typical hearts stained with triphenyltetrazolium chloride (TTC) from the study. The top panel shows an untreated heart (82% infarction), the middle a ticagrelor-treated heart (81% infarction), and the bottom an ischemically preconditioned heart (37% infarction). The tan tissue is TTC-negative and thus necrotic. The thin yellow or blue outlines mark the necrotic zones selected by Image-J’s color threshold function. The white rectangle at the bottom is a 2-cm wide standard.
Discussion
Previous investigations by us 2 –4 and others 1,5,8 have demonstrated the direct cardioprotective properties of 1 or more of the P2Y12 antagonists clopidogrel, ticagrelor, and cangrelor. We have found that the protective effect of cangrelor is dependent on the presence of platelets. Thus, in severely thrombocytopenic rats 9 and in isolated hearts perfused with cell-free, crystalloid buffer, 4,12 cangrelor no longer results in smaller infarcts. We now extend this observation to a second P2Y12 antagonist, ticagrelor. Although in the absence of platelets ticagrelor is no longer cardioprotective, its proposed ability to block ENT 1 should have been intact. Thus, it would appear that ticagrelor’s effect on adenosine uptake by itself is not sufficient to trigger protection in the isolated rat heart.
Cangrelor and ticagrelor are equally cardioprotective in our open-chest rat model, 2 but cangrelor has little effect on ENT 1. 7 The major metabolite of cangrelor does have a weak affinity for ENT 1, but the IC50 for inhibition of adenosine uptake in Madin-Darby canine kidney cells expressing human ENT 1 is 7-fold lower for ticagrelor than that for cangrelor metabolite, 7 and, therefore, ENT 1 inhibition would not be expected from an antiaggregation dose of cangrelor. Neither clopidogrel nor its active metabolite has any influence on ENT 1. 7 Clopidogrel and cangrelor were also similarly protective in our rabbit heart I/R model when clopidogrel was allowed enough time to achieve platelet inhibition equal to that from cangrelor. 4
We used 2 doses of ticagrelor in the present study. The 3 µmol/L concentration was based on published dose–response curves of ticagrelor’s ex vivo effect on ENT 1 from rat kidney. 13 The 10 µmol/L concentration was based on the treated buffer’s ability to block platelets in a blood sample. The 10 µmol/L concentration was higher than the theoretical blocking dose for the P2Y12 receptor probably because some of the ticagrelor would have been bound by serum proteins in the blood and also because the buffer sample was diluted 100% in the aggregometer by an equal quantity of blood. Whatever the case, neither dose showed any protection.
Both Ye et al 1 and Vilahur et al 5 observed greater cardioprotective effects with ticagrelor than with clopidogrel and attributed the difference to the former’s ability to block adenosine uptake and increase tissue adenosine levels. Ye et al 1 administered clopidogrel and ticagrelor intraperitoneally to rats only 5 minutes before reperfusion, but only ticagrelor was protective. As noted earlier, clopidogrel is a prodrug and must be enzymatically activated by a slow, multistep process by hepatic p450 enzymes. 14 Therefore, it is not surprising that there was no reduction in infarct size in their clopidogrel group since little of the active metabolite would have been present during the critical first few minutes of reperfusion. We found cangrelor to be highly protective when administered to rabbits 5 minutes prior to reperfusion, but it offers no protection when started just 10 minutes after reperfusion indicating P2Y12 receptor inhibition prevents a lethal injury that occurs in the first several minutes of reperfusion. 4 It is important to note that the effect of clopidogrel as well as ticagrelor on platelet aggregation was not tested by Ye et al 1 until the termination of the experiment 2 hours later, enough time for much of the clopidogrel to be converted to its active metabolite.
When clopidogrel was administered to pigs 4 hours before coronary occlusion, it was cardioprotective. But ticagrelor resulted in 23% smaller infarcts than clopidogrel, despite the fact that platelet inhibition during coronary occlusion based on aggregometry was not different between the groups and that the bleeding time was actually higher in the clopidogrel group. 5 The finding that the protection from ticagrelor could be partially blocked by the nonselective adenosine receptor blocker 8-(p-sulfophenyl)theophylline (8SPT) was offered as supportive evidence of the ENT-1 hypothesis. However, it was not tested whether 8SPT could block clopidogrel’s protection. 15
Prior investigations have demonstrated that many of the signaling steps triggered by P2Y12 antagonists that lead to cardioprotection are identical to those documented for conditioning interventions. We have shown this most extensively for cangrelor 4 but also for ticagrelor 3 and clopidogrel. 4 In the relevant signal transduction pathway, there are 2 points at which adenosine receptors are involved. 16 In the trigger sequence that occurs during the preconditioning ischemia, released adenosine can bind to A1 receptors to initiate the signaling. In the mediator phase that occurs at reperfusion, the A2B adenosine receptor is occupied because its affinity appears to be increased, presumably the result of phosphorylation by PKC. Both nonselective and selective A2B adenosine antagonists can block signaling and abort cardioprotection from pre- or postconditioning or the P2Y12 inhibitors cangrelor and clopidogrel. 4 Obviously, blockade of ticagrelor’s protection by a nonselective adenosine receptor antagonist can no longer be used as evidence that ticagrelor’s superior protection in pigs results from ENT 1-generated adenosine.
One shortcoming of our isolated rat heart model is that it does not include circulating red blood cells. Ticagrelor has been shown to promote release of adenosine triphosphate (ATP) from erythrocytes as well as inhibit adenosine reuptake by them. 17 Since ATP in the plasma would quickly be converted to adenosine, our model may lack an important adenosine source. Dipyridamole also blocks adenosine reuptake by erythrocytes, but its effect on infarct size after myocardial ischemia/reperfusion is confusing. Dipyridamole given acutely to dogs prior to in situ regional ischemia/reperfusion had no effect on infarct size at any of 3 doses tested 18 and 3 days of oral pretreatment of rats also had no effect. 19 On the other hand, acute administration of dipyridamole 5 minutes after onset of a 30-minute coronary occlusion in rats halved the infarct size. 20 Dipyridamole does, however, increase the potency of ischemic preconditioning against infarction in in situ rabbit hearts, but it only did so when administered prior to the short preconditioning period of ischemia. When started after the preconditioning ischemia but before the index ischemia, dipyridamole offered no significant protection indicating that blocking adenosine reuptake only increased ischemic preconditioning’s ability to trigger the protected state. 21 It is possible that ticagrelor’s blockade of adenosine uptake reinforces its preconditioning-like effect when given as a pretreatment as was done by Vilahur et al. 5 Because both cangrelor and ticagrelor were infused after ischemia had begun in our rat study, 2 those hearts might not have benefited from the adenosine boost. In the clinical setting of primary percutaneous intervention, such pretreatment with a platelet inhibitor would not be possible.
The present study failed to provide support for the hypothesis that augmented adenosine release from ticagrelor’s effect on ENT 1 is responsible for ticagrelor’s greater potency against infarction. But it does not disprove it either. A small increase in adenosine by ENT-1 inhibition may not by itself be enough to reach the threshold for infarct size reduction directly in the blood-free isolated heart. However, it is still possible that adding adenosine to the cardioprotective process resulting from platelet P2Y12 receptor blockade in an in situ heart could have augmented the protection. We cannot explain why ticagrelor is so much more protective in the pig than clopidogrel, but since no difference in potency was found between cangrelor and ticagrelor in our recent rat study, 2 we suspect either a possible species difference that may or may not involve adenosine or perhaps a protocol difference as discussed earlier.
Regardless of whether adenosine plays a role in its protection, we agree that in the setting of reperfusion therapy for acute myocardial infarction ticagrelor is a superior drug compared to clopidogrel. The latter suffers from an onset of effect that is too slow for acute treatment as well as a common genetic variation that negates its metabolic activation in a significant portion of the patient population. 22
The importance of the present study is underscored by controversies in the literature regarding the proposed mechanism of action of P2Y12 drugs solely as anti-platelet aggregants and a failure to fully appreciate their pleiotropic cardioprotective effects. Indeed, our recent work shows that drugs such as the caspase-1 inhibitor VX-765, which act via mechanisms distinct to the adenosine-dependent cardioprotective pathways, can significantly add to the cardioprotective effects of P2Y12 drugs that are delivered to patients with AMI as standard of care. 2 Together, ours and other studies suggest that all new anti-infarct interventions should be shown to be capable of adding protection to that from a P2Y12 inhibitor as part of the consideration for their clinical testing.
Footnotes
Authors’ Note
All applicable international, national, and institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by grant HL-118334 (DFA and JPA) from the Heart, Lung, and Blood Institute of the National Institutes of Health.