
Abstract
Platelet activation and aggregation during ischemia influence reperfusion-related myocyte necrosis, myocardial perfusion at the microvascular level, and thereby eventual recovery of cardiac performance. Inhibition of platelet activity therefore represents a worthwhile target to reduce cellular injury. The current study examined the effects of MK383 (tirofiban), a potent inhibitor of platelet aggregation, on infarct size and myocardial perfusion in canine subjects to either reocclusion (ie, 120-minute + 60-minute ischemia with intervening reperfusion) or prolonged occlusion (ie, 3 hours) followed by reperfusion (180 minutes). Platelet aggregation, infarct size (tetrazolium staining), coronary blood flow (flow probe), coronary vascular reserve, and myocardial perfusion (microspheres) were evaluated. MK383, administered at the time of reperfusion, produced a modest reduction of tissue necrosis (compared to saline-treated controls) in the reocclusion and prolonged occlusion studies. Blood flow in the infarct-related artery after coronary occlusion was comparable between treatment groups, as was myocardial perfusion in the deeper layers of the ischemic region; coronary vascular reserve decreased progressively during reperfusion. Of note, compensatory changes in blood flow within the adjacent nonischemic myocardium were not observed. In conclusion, we report that that limiting platelet aggregation during reperfusion impacted infarct development. Continued investigation into the mechanisms by which inhibition of platelet activity protects myocardium against ischemia–reperfusion injury and improves clinical outcomes is necessary.
Keywords
Introduction
Timely restoration of arterial patency (thrombolysis, angioplasty, etc) is an effective means to mitigate ischemic injury; however, complete reperfusion also triggers cellular pathways that exacerbate cardiocyte injury. The inability to reestablish organ perfusion at the microvascular level (ie, no-reflow) despite restoration of infarct-related artery patency is associated with poor prognosis independent of infarct size. 1 No-reflow is a dynamic process 2 –6 ; contributing causes include ultrastructural alterations at the level of the vascular endothelium, platelet aggregation, inflammation, embolization by atherosclerotic and thrombotic debris, and so on. 7 –10 Because of its potential role in organ healing and remodeling, the physiopathology of no-reflow and therapeutic strategies to counter its influence are the subject of ongoing investigation.
Platelet aggregation plays a crucial role in arterial thrombosis and is a contributing factor to postischemic reocclusion of an infarct-related artery and eventual clinical outcomes. 11 –14 Glycoprotein IIb/IIIa receptor inhibitors are useful for the management of no-reflow; for example, tirofiban reportedly limits no-reflow in animal models of infarction/reperfusion compared to either aspirin or clopidogrel. 15,16 We, 12 and others, 11,17,18 reported significant cardioprotection with these agents; however, the exact role that platelets play in myocardial infarction and no-reflow remains uncertain. Recent studies show that thienopyridines directly influence infarct size when administered just prior to reperfusion 19 ; their protective actions may include activation of different prosurvival signal transduction pathways. As such, these drugs are considered to act as postconditioning mimetics. 20,21 Accordingly, in the present study, we postulated that MK383, a potent glycoprotein IIb/IIIa inhibitor, would limit infarct-related cardiocyte injury principally by improving myocardial perfusion within the ischemic zone. Although the reocclusion model used here is uncommon, it does occur in patients who experience an earlier untreated acute cardiac event followed by partial spontaneous restoration of flow to the ischemic perfusion bed (ie, periprocedural myocardial infarction due to postsurgical or postinterventional revascularization 22 ). Additionally, we examined the potential preconditioning effect of a longer duration of ischemia and reperfusion prior to an extended second occlusion.
Materials and Methods
Dogs of either sex, weighing between 20 and 25 kg, were obtained from the Division of Laboratory Animal Services at Laval University; studies were carried out in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health (publication 85-23, revised 1996) and were approved by the Laval University Animal Ethics Committee.
Materials
MK383 (L-700462;
General Surgical Preparation
The anesthetic regimen and surgical preparation for these studies has recently been described. 23 Briefly, vascular introducer sheaths (8F; Terumo Medical Corp, Mississauga, Ontario, Canada) were inserted into the left and right femoral arteries; subsequently, a splenectomy was performed through a midline abdominal incision. 24 The heart was exposed via a left thoracotomy and instrumented; 2 cm of the left circumflex coronary (LCx—ischemic region) and left anterior descending (LAD—nonischemic region) arteries was dissected free of surrounding adventitial tissue and flow probes positioned. A snare occluder was placed immediately distal to the flow probe on the LCx artery (for coronary vascular reserve determinations and coronary occlusion). A catheter (#22 intracath) was inserted into the lumen of the LCx artery immediately distal to the snare occluder as previously described. 12,25 After placement of all catheters, heparin sodium (25 IU/kg, intravenous (IV)) was administered, the chest cavity covered (to prevent undue myocardial cooling), and dogs were allowed to stabilize for 30 minutes.
Aortic, coronary sinus, and coronary artery pressure catheters were connected to Gould P23XL pressure transducers. The micro-tipped pressure transducer (left ventricular (LV) pressure) was calibrated with systolic aortic and diastolic left atrial pressures. All hemodynamic measures including standard lead II of the scalar electrocardiogram and phasic coronary blood flow were recorded continuously (AxoScope; Molecular Devices Corporation, Sunnyvale, California) and stored on computer hard drive.
Experimental Protocol
Under steady state conditions, coronary vascular reserve in the LCx perfusion bed (posterior LV) was assessed: first by a 20-second acute occlusion and second (after return to steady state baseline conditions—∼5 minutes) by a single bolus injection of nitroglycerin (100 μg IC; SABEX, Boucherville, Quebec, Canada). After completion of these interventions, dogs were randomly assigned to either the saline or MK383 (300 µg/kg IV bolus followed by continuous infusion at 10 µg/kg/min) treatment groups. Saline or MK383 was administered starting 5 minutes prior to reperfusion; in the reocclusion study, dogs underwent 120-minute regional coronary occlusion (CO1) + 60-minute reperfusion (REP1) + 60-minute CO2 + 120-minute REP2. A bolus of xylocaine (10 mg IV; Astra Pharma, Inc, Mississauga, Ontario, Canada) was administered before coronary reperfusion to diminish the onset of ischemia- or reperfusion-induced arrhythmias. The experimental design is depicted schematically in Figure 1. During each reperfusion, coronary vascular reserve was assessed with nitroglycerin.

Schema of experimental protocols. In the reocclusion studies, dogs were exposed to 120-minute acute ischemia (CO1), 60-minute reperfusion (REP1), 60-minute acute ischemia (CO2), and 120-minute reperfusion (REP2). Platelet aggregation (venous blood from the coronary sinus) was assessed during the protocol (thick arrows). Reactive hyperemia (RH) and LCx responses to NTG were assessed at different time points indicated (long thin arrows). In the prolonged occlusion study, dogs underwent 180-minute ischemia and 180-minute reperfusion. In both studies, MK383 was administered intravenously starting 5-minute before reperfusion. Microspheres were injected at 4 time points (short arrows) to measure myocardial perfusion. LCx indicates left circumflex coronary; NTG, nitroglycerin.
Myocardial distribution of blood flow was determined at (1) baseline, (2) 30-minute CO1, (3) 30-minute REP1, and (4) 120-minute REP2 using microspheres suspended in saline (15 μm; NEN, Boston, Massachusetts). 12 At the end of the ischemia–reperfusion protocol, cardiac arrest was induced under deep pentobarbital anesthesia and infarct and risk regions delineated as previously described. 12
Hearts that fibrillated during either ischemia or reperfusion were cardioverted (DC shock ≤50 J) with paddles directly placed on the heart; in hearts that could not be defibrillated after 2 attempts, the animal was euthanized and the experiment terminated (data from these animals were not used in the statistical analysis).
To rule out the possibility of ischemic preconditioning bias by the reocclusion protocol, additional dogs were subject to prolonged coronary occlusion (3 hours) followed by reperfusion. Surgical preparation and treatment schedules were the same as described above (coronary vascular reserve and platelet aggregation/activation were not evaluated). Myocardial blood flow distribution was measured at (1) baseline, (2) 30-minute CO, (3) 30-minute REP, and (4) 180-minute REP.
Platelet Activity: Aggregometry and Flow Cytometry
Blood was collected into Vacutainer tubes containing 0.5 mL of 3.8% sodium citrate at (1) baseline, (2) REP1-60, (3) REP2-60, and (4) REP2-120. Platelet aggregation was assessed by whole blood aggregometry (model 592; Chrono-Log Corp, Havertown, Pennsylvania) using collagen (5 µg/mL; Chrono-Log Corp). 13 Samples were also analyzed within 2 hours of collection in a Coulter EPICS Elite ESP flow cytometer using a modification of the method described by Jy and coworkers 26 and as described in an earlier report from our laboratory. 12
Infarct Size, Myocardial Blood Flow, and Coronary Vascular Reserve Analysis
The LV was sliced into 5-mm-thick sections; the outline of each slice (ie, total LV area), necrotic area (AN), and the anatomic area at risk (AAR) were traced onto acetates for the determination of infarct size as described. 23 From the same slices, myocardial blood flow was obtained from the central portion of the nonischemic and ischemic zones; gamma activity was measured in a gamma-well scintillation counter (Cobra II, Canberra Packard Instruments, Montreal, QC, Canada.).
Baseline (Qbase) and peak (Qpeak) flow in the LCx perfusion bed after 20-second coronary occlusion and bolus nitroglycerin was assessed; coronary vascular reserve (ie, quotient of Qpeak/Qbase) was calculated from strip chart recordings. 27,28
Data Analysis
Differences in cardiac hemodynamics, phasic, and myocardial blood flow were determined by analysis of variance (ANOVA) and multiple comparisons were performed using the Student-Newman-Keuls multiple range test. The relation between infarct size and myocardial blood flow was analyzed using a multivariate analysis; infarct size (normalized to risk area) was the dependent variable, and blood flow in the subendocardial tissue layer of the ischemic zone (ie, coronary collateral flow) was the covariate. A probability (P) level of ≤.05 was considered statistically significant. Sample size necessary to obtain statistically valid results was determined before random assignment to the respective study groups 29 and was based on earlier studies from our laboratory with similar objectives 23,30 : 90% power to detect, at P ≤ .05, a minimum 20% change (expected standard deviation of 8%) in infarct size (treatment effect).
Results
Forty-one dogs entered the study; 7 dogs in the reocclusion study (5 saline and 2 MK383) could not be resuscitated after 2 attempts at cardioversion and succumbed to ventricular fibrillation; 20 dogs (n = 10 per group) survived the experimental protocol. In the prolonged occlusion study, 14 dogs completed the experimental protocol.
Platelet Aggregation and Activation
Effects of MK383 on platelet aggregation are summarized in Table 1. Flow cytometry of CD41 (a pan-platelet marker) activation in platelet-rich plasma is shown in Figure 2; MK383 effectively blocked spontaneous platelet aggregation.
Summary of Whole Blood Aggregometry Data.a

a Data are expressed in Ohms. REP1 indicates reperfusion period 1 at 10 or 60 minutes; REP2 indicates reperfusion period 2 at 60 or 90 minutes. Data are presented as mean (SD).
b P = 0.001 versus baseline values.

Effects of MK383 on collagen-induced platelet GP IIb/IIIa receptor activation. Platelet-rich plasma (venous blood from ischemic zone) was prepared as described (cf section “Methods and Materials”); spontaneous platelet activation (measured during reperfusion and compared to baseline) was markedly reduced after MK383 administration. GP indicates glycoprotein.
Cardiac Hemodynamics
For the reocclusion studies, baseline heart rate, LV, LCx perfusion pressure, and coronary sinus pressure were comparable between treatment groups; temporal changes are summarized in Table 2. Heart rate increased after CO1 and remained higher than baseline levels (P = 0.244). Rate-pressure product was the same in both groups, suggesting that myocardial oxygen demand did not affect infarct development. In the prolonged occlusion study, cardiac hemodynamics were also comparable for both treatment groups (cf Table 3).
Cardiac Hemodynamics for Reocclusion Studies.a,b

Abbreviations: CorSd, coronary sinus pressure during diastole; CorSs, coronary sinus pressure during systole; LCxPd, coronary artery pressure in the LCx artery during diastole; LCxPs, coronary artery pressure in the LCx artery during systole; LVPd, left ventricular pressure during diastole; LVPs, left ventricular pressure during systole.
a Data are presented as means (SD); n = 10 dogs per group.
b CO1, CO2 indicate coronary occlusion; REP1, REP2 indicate reperfusion periods. Time points (in minutes) are also indicated for the specific ischemia or reperfusion interventions.
Cardiac Hemodynamics for Prolonged Ischemia Studies.a

Abbreviations: AoPd, aortic pressure during diastole; AoPs, aortic pressure during systole; CorSd, coronary sinus pressure during diastole; CorSs, coronary sinus pressure during systole; CO-60, 60-minute coronary occlusion; LVPd, left ventricular pressure during diastole; LVPs, left ventricular pressure during systole; REP-60, indicates 60-minute reperfusion; REP-180, 180-minute reperfusion.
a Data are means (SD); n = 7 dogs per group.
Coronary Vascular Reserve and Blood Flow
Coronary vascular reserve (not done in prolonged occlusion study) was similar in saline and MK383 groups (Table 4), and no change in regional blood flow occurred in the adjacent perfusion bed during hyperemia. Responses to bolus nitroglycerin (ie, endothelium-independent vasodilator) included a significant transitory increase in LCx flow under control conditions; however, coronary vascular reserve progressively decreased during reperfusion (Figure 3). Note that blood flow remained constant in the anterior perfusion bed during nitroglycerin administration, thus confirming the lack of a washout effect between perfusion beds. Phasic LCx blood flow (measured with flow probe) for the reocclusion protocol is shown in Figure 4; LAD artery blood flow was constant for the duration of the experiment.
Summary of Reactive Hyperemia and Intracoronary Bolus Nitroglycerin Responses.a

Abbreviations: LAD, left anterior descending; LCx, left circumflex coronary; SD, standard deviation.
a Data are presented as means (SD).
b P = .01 versus baseline values.
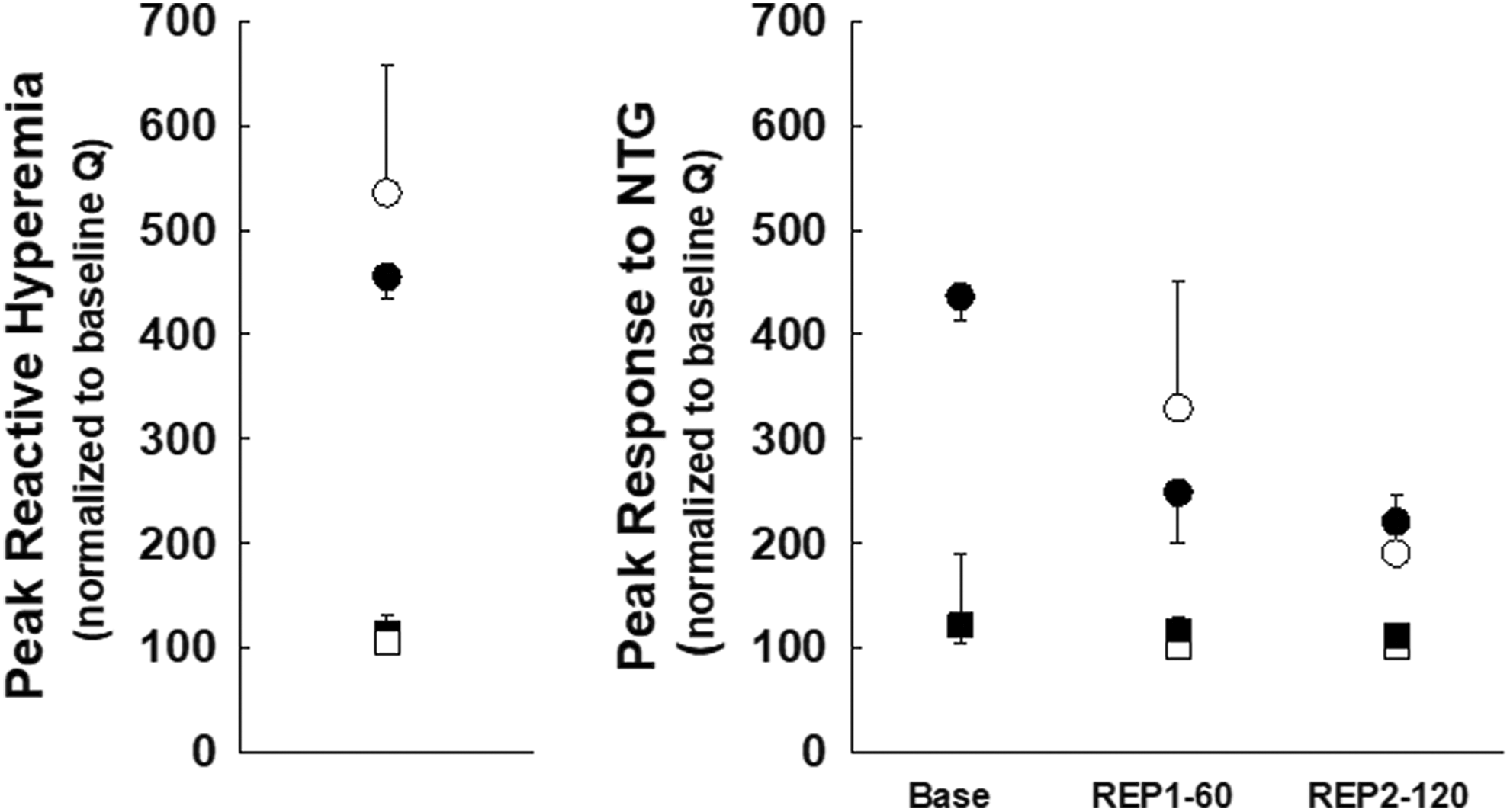
Peak reactive hyperemia responses to 20-second occlusion in the infarct-related artery (ie, LCx) of saline- (closed spheres) and MK383- (open spheres) treated dogs (left panel). Responses to intracoronary nitroglycerin are shown (right panel) at baseline and during REP1 (60 minutes) and REP2 (120 minutes); data are means (SD). Reactive hyperemia responses and vessel reactivity with nitroglycerin in the nonischemic zone (ie, LAD artery) for saline (open squares) and MK383 (filled squares) are shown. LAD indicates left anterior descending; LCx, left circumflex coronary; SD, standard deviation.

Phasic coronary blood flow (mL/min; electromagnetic flow probe) changes for the ischemic zone (ie, LCx artery; upper panel) in saline (filled spheres) and MK383 (open spheres) groups; the lower panel shows phasic coronary blood flow for the nonischemic (ie, LAD artery) region. There were no significant differences between groups; results are means (SD). LAD indicates left anterior descending; LCx, left circumflex coronary; SD, standard deviation.
Myocardial perfusion across the LV wall in ischemic and nonischemic regions for both studies (ie, reocclusion, prolonged ischemia) is shown in Figure 5; at REP1 and REP2, endocardial layer blood flow (mL/min/g) within the area at risk trended to higher but not significant levels compared to baseline (perfusion in nonischemic myocardium was not affected).

Myocardial perfusion (ie, microspheres) across the LV wall in the ischemic (upper panel) and nonischemic (lower panel) beds of saline (black bars) and MK383 (gray bars) groups at baseline, coronary occlusion, REP1, and REP2 in dogs from the reocclusion (left panels) and prolonged ischemia (right panels) studies. Data are means (1SD). LV indicates left ventricular; SD, standard deviation.
Infarct Size
In the reocclusion study, AAR (% of LV area) was approximately 45% in saline and MK383 groups (cf Figure 6); infarct size was 36.0% (7.0%; data are mean [standard deviation]; ANOVA) versus 29.7% (10.2%; P = .124), respectively, without consideration of individual variations in coronary collateral blood flow. Collateral blood flow (ie, subendocardial layer blood flow) during 120-minute occlusion period was not affected by treatment with MK383. Collateral blood flow is a baseline predictor of infarct size 31,32 ; an inverse relationship exists between collateral blood flow and infarct size (ie, larger infarcts with low flow and vice versa). 33,34 This relationship was found in dogs subjected to the reocclusion protocol; the relation shifted downward (P = 0.014; mean likelihood ratio) in MK383 dogs suggesting a cardioprotective effect but protection was not caused by improved perfusion within the ischemic myocardium.

Risk zone size (as % LV area) and AN/AR (as % AR) for saline (closed bar) and MK383 (open bar) groups for dogs exposed to the reocclusion protocol. Results are means (SD). The relation between myocardial infarct size and collateral blood flow within the area at risk is illustrated. Results for saline (filled circles; y = 47.5 − 137.8x, r2 = 0.75) and MK383 (open circles; y = 37.7 − 122.2x, r2 = 0.27) groups are shown; each data point represents an individual dog. AN indicates necrotic area; AR, area at risk; LV, left ventricular; SD, standard deviation.
In the prolonged occlusion study, the AAR was approximately 40% of LV area in both groups (Figure 7); infarct size was 33.3% (7.0%) versus 23.7% (6.7%; P = 0.053; ANOVA) in saline and MK383-treated dogs. A parallel, downward shift of the collateral blood flow/infarct size relation (P = 0.012; mean likelihood ratio) indicates reduction of infarct size by MK383.
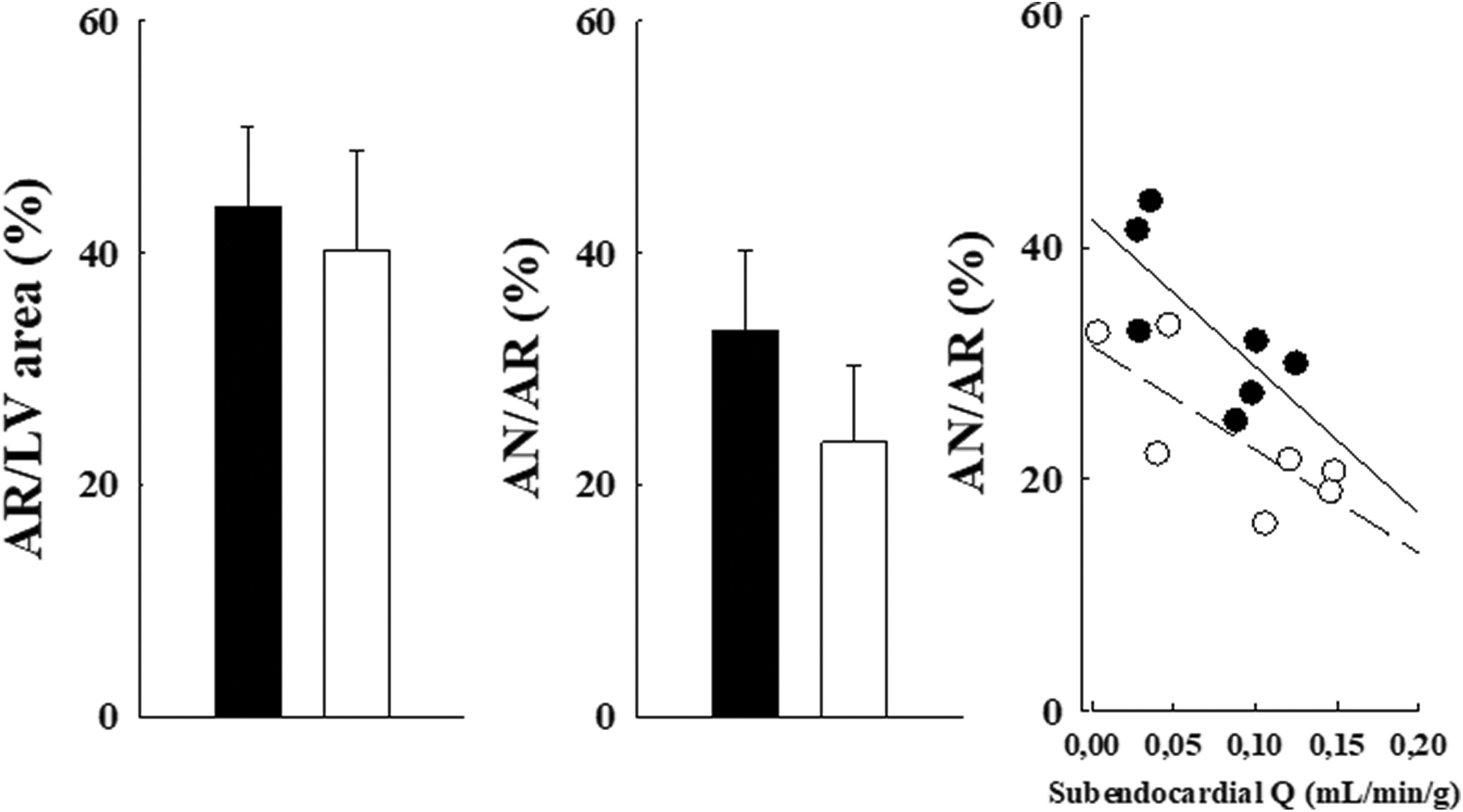
Risk zone size (as % LV area) and AN/AR (as % AR) for saline (closed bar) and MK383 (open bar) groups in dogs exposed to prolonged occlusion. Results are means (SD). The relation between myocardial infarct size and collateral blood flow within the area at risk is illustrated. Results for saline (filled circles; y = 42.4 − 126.7x, r2 = 0.52) and MK383 (open circles; y = 31.5 − 89.2x, r2 = 0.58) groups are shown; each data point represents an individual dog. AN necrotic area; AR, area at risk; LV, left ventricular; SD, standard deviation.
Discussion
We document that MK383, administered prior to coronary reperfusion, was able to mitigate ischemic injury; cardioprotection (ie, ±15% reduction) was only observed when myocardial perfusion within the ischemic region was considered in the statistical analysis. Findings were not due to improvements in cardiac hemodynamics, myocardial perfusion, or a lowering of myocardial oxygen demand. We also questioned whether ischemic conditioning could account for protection (in the reocclusion study); however, results do not support this hypothesis. Earlier studies have documented that protection mediated by classical preconditioning is lost in dogs when acute coronary occlusion is extended to 3 hours. 31
Role of Platelet Activation
Platelets, and their multiple interactions with different cell types, represent a key therapeutic target to limit ischemic injury. Restoration of arterial patency stimulates an inflammation cascade that releases pro-inflammatory cytokines that promote cardiac dysfunction and aggravate myocardial injury. 35 Potent chemotactic factors are released by activated platelets that promote thrombus formation and microaggregation, 36 resulting in obstruction of microvasculature and disruption of tissue perfusion. 17,28,37 Platelets can also modulate chemotactic properties through platelet–leukocyte and platelet–endothelium interactions 38 ; neutrophils, oxygen-free radicals, and edema formation are considered important though secondary participants. 39 –41
Myocardial Blood Flow and No-Reflow
Timely restoration of coronary blood flow to ischemic myocardium improves cardiac performance and clinical outcomes 42 ; however, restoration of epicardial vessel patency postischemia does not necessarily guarantee optimal conditions for ventricular performance. Structural vascular remodeling also affects vascular tone and circumferential wall tension, which affect metabolic demand and blood supply equilibrium and influence collateralization 43 and microvessel rarefaction. 44,45 All of these factors contribute to the so-called “no-reflow phenomenon” that is associated with LV dilatation and remodeling 7,15,46 and poor prognosis. 47
Infarct Size
Although platelet activation causes myocardial damage, 48 it remains unclear that poor cellular survival is caused by either platelet accumulation or release of platelet-derived compounds. A host of antiplatelet therapies are currently being used in clinical practice (cf reviewed in the study by Michelson 49 ) to prevent no-reflow and protect against ischemic injury. 13 In patients, combined tirofiban plus heparin (ie, PRISM-PLUS) reduced thrombus burden and improved microvascular perfusion to the region. 50 –52 Kloner recently provided critical discussion of platelet inhibition and cardioprotection 53 ; antiplatelet therapies, in general, were considered to provide some benefit against ischemic injury, but physiopathological mechanisms remain unclear. In most, but not all, animal studies, protection against necrosis and arrhythmogenesis with platelet-activating factor blockers is reported. 12,13,48,54 –56 In the present study, MK383 administered at the time of reperfusion reduced infarct size; protection only reached a level of significance when collateral blood flow within the AAR (ie, not detected by ANOVA) was considered in the statistical analysis; the importance of collateral blood flow as an independent predictor of infarct size has previously been discussed. 33,34,57 Infarcts in the reocclusion study were approximately 15% smaller with MK383 (compared to saline group); protection was also observed in the prolonged occlusion (ie, 3 hours) study. The latter results confirm earlier findings that protection is possible with longer durations of ischemia 15 ; however, our findings differ somewhat in that we did not observe a greater degree of myocardial perfusion. This may be due to the different methods used to measure blood flow in the deeper myocardial layers (ie, microspheres vs contrast echocardiography). Lack of effect (by MK383) on myocardial blood flow suggests a direct protective effect of the drug on heart muscle. Preconditioning per se in the reocclusion study does not appear to affect infarct development; however, recent findings suggest that different antiplatelet agents (ie, tirofiban, P2Y12 blockers) administered at the time of reperfusion act as postconditioning mimetics. 58 In addition, tirofiban enhances production of nitric oxide 17,18,59 within ischemic myocardium to mitigate endothelial dysfunction at the microvessel level; it also reduces inflammatory exudation, thereby limiting structural damage. MK383 also reportedly stimulates/activates different intracellular signaling pathways known to be involved in cardiac postconditioning 19,20 ; however, the experimental protocol used here is not postconditioning in the classical sense. Other factors pertinent to no-reflow that may be influenced by tirofiban (or similar acting agents) include blockers of platelet–neutrophil interactions and tissue factor activity 60,61 ; blockade of platelet adhesion could reduce production of oxygen radicals, which exacerbate tissue injury during the early phase of reperfusion. 62
Limitations
Risk factors such as duration, depth of ischemia, and AAR affect infarct development and were evaluated in each animal. 63,64 Gender also affects cardiac sensitivity to ischemic injury 65,66 but was not considered as an independent variable of infarct development in the current study (11 female dogs used here were randomly assigned to the different groups, each of which comprised at least 2 females). Myocardial injury was performed in healthy animals; however, it is understood that clinical relevance necessitates consideration of the effects of various comorbidities on infarct development.
Herein we did not directly measure no-reflow; perfusion deficit in the deeper myocardial layers was based on microsphere data. Duration and severity of ischemia–reperfusion injury is also an important consideration for development and expansion of no-reflow and potential treatment effects. 67,68 Brief episodes of ischemia–reperfusion produce significant no-reflow in different experimental models 2 –4 ; however, no-reflow is more extensive when occlusion and reperfusion are ≥3 hours duration. 7 Cardioprotection with MK383 was only established when collateral blood flow was incorporated as a covariate in the statistical analysis 69,70 ; a legitimate explanation could be that these studies were underpowered to demonstrate a salutary treatment effect. 71 However, we followed the recommendations for rigor and reproducibility described in the Animals in Research: In vivo Experiments guidelines. 72 Sample size determination and power were established prior to randomization of animals to the respective study groups; considering the inclusion of known determinants of infarct size for the canine model of ischemia–reperfusion injury, we believe that group “n” values were within established norms. 15,23,73
Conclusions
In the current study, MK383 administered prior to coronary reperfusion reduced myocardial injury; results also confirm the importance of measuring coronary collateral blood flow. Potential benefits of ischemic conditioning (in addition to MK383 treatment) were not observed; however, we cannot rule out the possibly that MK383 stimulated intracellular survival pathways that are associated with postconditioning-mediated protection. We also show an important loss of coronary vascular reserve that was not altered by MK383; these findings suggest a limited capacity to preserve vascular integrity. Of note, greater perfusion of the adjacent vascular bed (ie, anterior perfusion bed) during ischemia was not observed. Further investigation is required regarding infarct development and postischemic myocardial perfusion after restoration of arterial patency; cross talk between platelets and inflammatory cells may be a crucial physiopathologic mechanism that contributes to microvascular obstruction and poor prognosis.
Footnotes
Acknowledgments
MK383 was a generous gift from Merck, Rahway, New Jersey. The author would like to thank Denys Simard for helpful editorial commentary, Dany Patoine for assistance with GraphPad Prism, and Dr Marc Bossé for the flow cytometry analyses.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for these studies was obtained from the Heart and Stroke Foundation of Canada and the Québec Heart Institute.