
Abstract
Limiting the injurious effects of myocardial ischemia-reperfusion is a desirable therapeutic target, which has been investigated extensively over the last three decades. Here we provide an up to date review of the literature documenting the experimental and clinical research demonstrating the effects of manipulating cGMP for the therapeutic targeting of the injurious effects of ischemic heart disease. Augmentation of the cyclic nucleotide cGMP plays a crucial role in many cardioprotective signaling pathways. There is an extensive body of literature which supports pharmacological targeting of cGMP or upstream activators in models of ischemia-reperfusion to limit injury. NO donors have long been utilised to manipulate cGMP, and more recently non-NO synthase derived NOx species have been investigated, resulting in their evaluation in clinical trials for the treatment of ischemic heart disease. Encouraging results demonstrate that natriuretic peptides are worthy candidates in manipulating cGMP and its downstream effectors to afford cytoprotection. Synthetic ligands have been designed which co-activate natriuretic peptide receptors to improve targeting this pathway. Advances have been made in targeting the soluble guanylyl cyclase which catalyzes the production of cGMP independently of the endogenous ligand NO using NO-independent stimulators and activators of sGC. These novel compounds show promise as a new class of drugs that target this signaling cascade specifically under pathological conditions when endogenous NO production may be compromised. Regulating the degradation of cGMP via phosphodiesterase inhibition also shows therapeutic potential. It is clear that production and regulation of cGMP is complex, indeed its spatial production and cellular distribution are only just emerging.
Keywords
Enhancing the tolerance of myocardium to the lethal consequences of ischemia and reperfusion, especially myocardial necrosis (infarction), remains a highly desirable therapeutic goal. Although recent advances in primary prevention and timely reperfusion have reduced early mortality from acute myocardial infarction, long-term prognosis is directly related to the extent of tissue necrosis sustained during the index coronary occlusion/reperfusion insult. During the last 3 decades or so, sustained exploration of the endogenous physiological signaling cascades recruited to enhance tolerance has demonstrated that there are many components to the multifaceted conditioning paradigms (preconditioning and postconditioning). One such component is 3′,5′-cyclic guanosine monophosphate (cGMP), a cyclic nucleotide responsible for eliciting many potentially cardioprotective actions.
Historical Background
3′,5′-Cyclic guanosine monophosphate (cGMP) is an intracellular second messenger produced from the purine nucleotide guanosine triphosphate (GTP) in a reaction catalyzed by cytosolic soluble guanylyl cyclase (sGC) or membrane-associated particulate guanylyl cyclase (pGC). Following the discovery of 3′-5′-cyclic adenosine monophosphate (cAMP) in the late 1950s, 1 cGMP was confirmed to be a physiological mediator when Price et al 2 isolated it from rat urine in 1963 and Sutherland’s laboratory demonstrated that steroid, thyroid, and pituitary hormones affected urinary excretion of cGMP. Fifty years after its discovery, cGMP is now established as a ubiquitous second messenger, mediating a large variety of physiological processes.
Within the cardiovascular system, cGMP is well established as an important pharmacological target. Indeed, targeting cGMP signaling for the therapeutic treatment of cardiovascular disease is not a recent discovery. Lauder Brunton’s description of the effects of amyl nitrite in angina pectoris in 1867 and William Murrell’s subsequent description of the effects of glyceryl trinitrate (nitroglycerin) in 1879 preceded any understanding of the mechanism of action by more than a century. 3 Following the description of an (then) unidentified endothelium-derived relaxing factor (EDRF) in the early 1980s, 4,5 it was demonstrated that EDRF increases cGMP synthesis in isolated blood vessels. 6,7 The subsequent identification of EDRF as nitric oxide (NO) 8 and its association with guanylyl cyclase activity and cGMP synthesis 7 opened up an area of biology with huge potential for pharmacological exploitation, especially in the cardiovascular system. In the early 1990s, cGMP was shown to be an important cyclic nucleotide in affording the preconditioned state in rabbit myocardium. 9
In the late 1980s and early 1990s, the sequential discovery of a series of peptide mediators produced within the cardiovascular system, called natriuretic peptides (NPs), opened up a new dimension in cGMP signaling. The major members of this family—in order of discovery, atrial NP (ANP), brain- or B-type NP (BNP), and C-type NP (CNP)—all elicit their biological actions, at least in part, through elevation of intracellular cGMP. The NP membrane-associated receptors, NP receptor (NPR) A and NPR-B, contain guanylyl cyclase domains capable of catalyzing the conversion of GTP to cGMP. This pGC activity is, as far as we know, uniquely activated by the NP family of mediators that include ANP, BNP, CNP, and the related guanylins and atriopeptins. This further dimension to cGMP regulation in the cardiovascular system provides additional complexity and opportunities for therapeutic manipulation.
Over the last 30 years, extensive investigation has focused on the physiological roles and regulation of the cGMP pathway, its disruption in various diseases, and its therapeutic “druggability.” In this review, we explore the extensive literature focusing on myocardial cytoprotection against ischemia–reperfusion injury. We consider the basic research and consequent clinical studies that have followed with particular reference to the clinical treatment of acute myocardial infarction (AMI).
Production and Localization of cGMP
3′,5′-Cyclic guanosine monophosphate production is mediated by 2 distinct guanylyl cyclases that are spatially discrete within the cell. Particulate guanylyl cyclase is described as membrane associated and is confined to the plasma membrane and connected to the extracellular ligand-binding domain via a kinase homology region. Atrial NP shows the highest affinity for the NPR-A receptor, while BNP is 10-fold less potent. 10 C-type NP is the primary ligand for NPR-B, stimulating guanylyl cyclase with 50- to 500-fold greater affinity than ANP and BNP (Figure 1). 11 Soluble guanylyl cyclase is found in the cytoplasmic domain, catalyzing the production of cGMP primarily through the binding of the endogenous ligand NO. Soluble guanylyl cyclase is a heterodimeric heme protein consisting of both α- and β-subunits. To date, 2α isoforms and 2β isoforms have been identified. 12,13 The α1β1 protein has been most extensively researched and is found in most tissues, including the kidney, brain, heart, and vascular tissue. 14 A heterodimer consisting of both subunits is essential to form the catalytically active sGC. 15,16 The N-terminal histidine 105 heme-binding site of the β subunit forms the basis of the NO-sensing region, which when occupied by NO leads to a 200-fold increase in the synthesis of cGMP (Figure 1). 17 Endogenous CO has also been shown to activate sGC but producing only a 4-fold increase in activity. 18

Structural representation of natriuretic peptide receptors (NPRs) and soluble guanylyl cyclases (sGCs) implicated in the production of cGMP. NB: NPR-C does not have particulate guanylyl cyclase activity and its mechanism of possible initiation of cGMP production remains to be eluded. Soluble guanylyl cyclase exists in 2 redox states that can catalyze cGMP production, however the oxidized form of sGC has no endogenous ligand. cGMP indicates 3′,5′-cyclic guanosine monophosphate.
3′,5′-Cyclic guanosine monophosphate activity is governed by specific cGMP-binding motifs on target proteins and its highly regulated degradation by phosphodiesterases (PDEs). 3′,5′-Cyclic guanosine monophosphate-dependent protein kinase (PKG) represents the primary cGMP mediator, which phosphorylates target proteins. 3′,5′-Cyclic guanosine monophosphate-dependent protein kinase I shares its allosteric cGMP-binding motif with 3′,5′-cAMP-dependent protein kinase (PKA; 50-fold lower selectivity than cAMP) and a second cGMP effector, the cyclic nucleotide-gated (CNG) ion channel. The second binding motif occurs only in the cGMP-regulated PDEs. Indeed, it is this complex combination of effectors that tightly regulates cGMP signaling. The PKG, PKA, and CNG channels are colocalized with effector proteins in different cell types, resulting in differing responses to elevated cGMP levels. For example, PKG is colocalized with phospholamban (PLB) and the inositol trisphosphate (IP3) receptor in the sarcoplasmic reticulum in smooth muscle cells. In contrast, PKA is colocalized with PLB and IP3 in cardiac myocytes eliciting differing responses to elevation of cGMP. Disruption of cGMP signaling has been identified in many aspects of cardiovascular function and disease including platelet aggregation, inotropy, cell growth/proliferation, and apoptosis, all of which rely on tightly regulated and specific signaling.
Hydrolysis of cGMP to 5′-GMP occurs through PDE catalytic activity. There are 10 families of mammalian PDE, 4 of which hydrolyze both cGMP and cAMP (1, 2, 3, and 10) and 3 which are specific for cGMP (5, 6, and 9). Specific targeting sequences found in PDEs traffic them to specific proteins leading to their being expressed in discrete subcellular locations. The net effect of this is that the intracellular concentration of cGMP varies within different subcellular compartments. Of the PDE isozymes, PDE2 and PDE5 are of particular interest because they have been demonstrated to show distinct regulation of specific cGMP subcellular concentrations as a result of pGC and sGC-mediated production, respectively. 19 Thus, there is a multifaceted approach to regulating cGMP and its downstream effectors. In this review, we will focus on potential therapeutic targets that can augment the production, distribution, localization, and metabolism of cGMP to limit the cytotoxic effects of myocardial ischemia–reperfusion injury, resulting in myocardial necrosis.
Nitric oxide, Nitrite, and Nitrate
Nitric Oxide
Nitric oxide is a gaseous autacoid formed by the action of NO synthase (NOS) on
Nitrate and Nitrite as Potential NO Reserves
An alternative pathway for NO formation involving nitrate (NO3 −) and nitrite (NO2 −), which predominates when tissue oxygen tension is low, has been described in mammals. 25 Nitrate and NO2 − can be considered reservoirs for NO when NOS-dependent NO levels are insufficient to maintain cellular homeostasis. 21 Nitrate and NO2 − can be reduced in sequential steps by enzymatic and nonenzymatic mechanisms creating new pools of NO with the potential to activate the sGC/cGMP/PKG pathway or by other processes that are distinct from this classical route of intracellular signalling. 21 The reaction of NO with oxyhemoglobin leads to the production of NO3 − and the formation of methaemoglobin. 26,27 Green leafy vegetables are a major source of NO capable of delivering more NO3 − than all the NOS isoforms combined. 21,28 Nitrite is formed following autooxidation of NO, nonenzymatically by deoxyhemoglobin 29 and by the enzymes ceruloplasmin, xanthine oxidoreductase, and aldehyde oxidase. 30 The mitochondrion has been shown to be a source of NO2 − and to exhibit NO2 − reductase activity. 31 Plasma NO3 − concentration in humans is approximately 2 orders of magnitude greater than that of NO2 − ranging from 20 to 50 μmol/L 32 ; plasma NO2 − concentrations in fasting humans are typically within the range of 50 to 500 nmol/L. 33 –36
Both NO3 − and NO2 − were considered terminal products of NO synthesis but are now recognized as reserve sources for NO production, especially under conditions of hypoxia. 31 Nitric oxide derived particularly from NO2 − has been shown to modify proteins and redox state, 37 which may be determinant factors in facilitating cytoprotection in models of ischemia–reperfusion injury in the heart and other organs. 38 The precise mechanism by which NO2 − is reduced to NO or whether NO is released from erythrocytes to promote vascular smooth muscle relaxation is currently unclear. 31 Nitrite-derived NO 39 and dinitrogen trioxide (N2O3) 40 have been proposed as nitrogen oxide species responsible for promoting NOS-independent vasodilatation in humans. Lundberg and colleagues 21 suggest that both species form in the circulation, the former liberated by the reaction of NO2 − with deoxyhemoglobin and the latter from the reaction of NO2 − bound methemoglobin and NO, forming a nitrogen dioxide radical, which then reacts with free NO to form N2O3.
Nitrate, NO2 −, and NO Signal Transduction Mechanisms: cGMP Versus S-Nitrosylation
The mechanisms by which NO3 − and NO2 − are converted to NO have already been highlighted; it is presumed that this pool of NO is capable of activating the same intracellular pathways as NOS-derived NO. Nitric oxide stimulates sGC leading to the generation of cGMP, thereby affecting downstream mechanisms that modulate cell function. This classical form of NO signaling has been discussed extensively in comprehensive reviews. 20,41 Nitric oxide derived from inducible NOS (iNOS) has a pertinent role in regulating and contributing to signaling associated with oxidative stress in pathological states including inflammation. However, the numerous intricate mechanisms and processes involved are beyond the scope of this review. We would refer readers to excellent reviews by Pfeilschifter et al, 42 Danson et al, 43 and Klein 44 for a fuller examination.
There is growing interest in another form of NO signaling independent of sGC. This involves modification of protein thiol groups. S-Nitrosylation implies the binding of NO to cysteine residues, altering the stability, binding, activity, localization, and irreversible oxidation of cysteine rich proteins, which are numerous. 37,45,46 For example, S-nitrosylation increases sarcoplasmic/endoplasmic reticulum adenosine triphosphatase (ATPase; SERCA) activity, 47 increases connexin 43 gap junction communication, 48 and inhibits phosphatase and tensin homolog activity 49 preventing inhibition of phosphoinositide-3-kinase/protein kinase B (PI3K/Akt), which are significant factors in the context of myocardial ischemia–reperfusion and cardioprotection. 37 Ischemic preconditioning effected by brief periods of ischemia and reperfusion prior to a sustained ischemic insult is cardioprotective. Mouse hearts subjected to this form of cardioprotective intervention were found to have increased S-nitrosylation of L-type calcium channels and SERCA decreasing intracellular calcium load, and S-nitrosylation of the F1 ATPase-limiting adenosine triphosphate (ATP) breakdown. 47
The Cytoprotective Action of NO2 −
Organic and inorganic NO3 − and NO2 − therapy has many beneficial effects in the cardiovascular system. Numerous studies during the last 2 decades have examined the protective actions of organic NO3 −s and other NO donor compounds in models of cardioprotection against ischemia–reperfusion injury and have demonstrated the ability of the acutely administered agents to limit infarct size. Preliminary data have demonstrated that nitroglycerin can limit infarct size in isolated rat hearts when administered during early reperfusion. More recently, however, the potential of inorganic NO2 − and naturally occurring dietary NO2 − as cardioprotective sources of NO has been examined. Sodium nitrite (10-100 μmol/L) reduced infarct size by some 70% in the rat isolated heart. 51 The protective effect of NO2 − given for the full duration of global ischemia but not reperfusion was reversed by the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, suggesting that NO2 −-mediated cardioprotection is in part NO dependent. 51 Nitrite cardioprotection appears to be NOS independent as it was shown that sodium nitrite limitation of infarct size in a mouse model was still present during pharmacological NOS inhibition or in eNOS knockout mice. 52 Other studies implicate xanthine oxidase activation and KATP channel opening in sodium nitrite-induced infarct size limitation in the rat heart in vivo and ex vivo. 51,53 There is evidence to suggest that NO2 −-derived NOS nitrosylates mitochondrion complex 1 during ischemia–reperfusion, delaying mitochondrial permeability transition pore (mPTP) opening, ROS formation and cytochrome c release, and ATP depletion, thus conferring cardioprotection. Shiva and colleagues 54 found that sodium nitrite given 24 hours prior to ischemia or acutely prior to reperfusion caused a marked reduction in infarct size in mouse hearts. Furthermore, it was demonstrated that sodium nitrite improves mitochondrial respiration following hypoxia in a concentration-dependent manner and delayed mPTP opening and cytochrome c release in isolated mitochondria subjected to hypoxia and reoxygenation. 54 In contrast to these data, it has been demonstrated that NO can inhibit cytochrome c oxidase (complex IV) activity, in mitochondria isolated from rat brain 55 and skeletal muscle 56 following incubation with sodium nitroprusside and S-nitrosoglutathione, respectively.
Bryan and coworkers 57 found that mice given free access to chow and water supplemented with sodium nitrite for 7 days were more resistant to myocardial ischemia–reperfusion injury, compared to mice on a standard diet, displaying a 48% relative reduction in infarct size compared to mice that did not receive sodium nitrite. 57 Furthermore, dietary sodium nitrite significantly increased plasma and tissue nitrosylation products, for example, S-nitrosothiols and nitrosoproteins, increased steady state concentrations of NO3 −, NO2 −, and nitrosyl heme products. 57 These hallmark features are likely to be very important in conferring NO2 − protection of ischemic myocardium. Most recently, it has been demonstrated that acute ingestion of dietary NO3 − in the form of beetroot juice elevated circulating NO3 − and NO2 − levels in humans and elevated platelet cGMP levels in healthy males only. 58 Although this evidence is not associated with cardiovascular cytoprotection, it highlights the potential for dietary or supplemented NO2 − as a credible mechanism for cGMP elevation. Previously, the authors demonstrated that beetroot juice lowered blood pressure in healthy volunteers, elevating plasma NO2 − and cGMP levels. 59
Pluta and colleagues 60 examined the safety and feasibility of long-term NO2 − therapy in healthy volunteers administered sodium nitrite intravenously (iv) for 48 hours. Cardiac NO3 −, NO2,− and S-nitrosothiol levels in plasma increased in all patients and returned to preinfusion baseline levels within 12 hours. The authors concluded that prolonged NO2 − iv infusion below the maximal tolerated dose is safe for use in treatment of major diseases including ischemic heart disease. Side effects were limited to asymptomatic transient decreases in arterial blood pressure and asymptomatic increases in methemoglobin. Indeed, the Nitrites In Acute Myocardial Infarction (NIAMI) trial is a placebo-controlled, double-blind, phase 2 clinical trial that aims to determine whether a 5-minute iv infusion of sodium nitrite (14 μmol/mL) just prior to percutaneous coronary intervention can reduce infarct size and associated enzyme biomarkers including creatine kinase and troponin I in patients presenting with ST-segment elevation myocardial infarction (STEMI). 61 Other end points include left ventricular ejection fraction and end-diastolic volume at 1 week and 6 months after myocardial infarction (MI), and infarct size measured by magnetic resonance imaging at 6 months post-MI. 61 This multicenter clinical study is the first investigating whether sodium nitrite infusion can reduce myocardial infarct size in humans. This study may provide a significant platform for establishing sodium nitrite as first-line therapy in the clinical treatment of AMI.
Receptor-Mediated NOS Activation
Many receptors have been linked to PI3K/Akt/eNOS signaling and elevation of intracellular NO including bradykinin, adenosine, and opioid peptide receptors. Most recently, β3-adrenergic receptor (β3-AR) stimulation (see Figure 2) has been demonstrated to stimulate NO generation via eNOS in the left and right human myocardium, which is blocked in left ventricle (LV) by the PI3K inhibitor LY294002. 62 Aragon and colleagues 63 recently demonstrated that β3-AR stimulation limits infarct size when administered at reperfusion in mice. This protection was accompanied by an elevation in eNOS PSer1177 and elevated nNOS levels. Whether this protection is mediated by sGC/cGMP signaling or possible inhibition of mitochondrial respiration and xanthine oxidase 64 remains to be elucidated.

Schematic representation of cytoprotective signaling cascades in cardiac myocytes incorporating cGMP. Included is the incorporation of NO3 −/NO2 − as an alternative source/store of NO. Highlighted is the dynamic redox of sGC which is known to exist in vivo. Production of cGMP can be achieved by endogenous NO targeting the reduced sGC or exogenously by sGC stimulators. In addition, sGC activators can target the oxidized state. Elevation of cGMP can also be achieved by targeting the membrane-associated particulate guanylyl cyclase via the natriuretic peptide receptor ligands. 3′,5′-Cyclic guanosine monophosphate is depicted in spatially distinct areas produced by different guanylyl cyclases and regulated by specific phosphodiesterases. Cytoprotective signaling via cGMP has been demonstrated to culminate in inhibition of the mPTP via reduced intracellular calcium concentrations and inhibition of mKATP channels. cGMP indicates 3′,5′-cyclic guanosine monophosphate; NO3 −, nitrate; NO2 −, nitrite; NO, nitric oxide; sGC, soluble guanylyl cyclase; mPTP, mitochondrial permeability transition pore; mKATP, mitochondrial ATP-sensitive K channel.
Nitric Oxide-Independent Stimulation/Activation of sGC
In addition to elevating cGMP via manipulation of NO, targeting sGC directly has become tractable in recent years. In this section, we discuss the redox that sGC exists in, how the different redox states can be specifically targeted, and the possibility of utilizing CO as an alternative ligand.
Soluble Guanylyl Cyclase Redox
The rate of reaction under physiological conditions is regulated by diffusion; however, a complex redox equilibrium of sGC exists, identifying challenges in successfully targeting this pathway. It is now well accepted that sGC can exist in 3 different forms depending on the redox state of the central heme group. 65 The reduced (ferrous) heme group is critical for NO sensing and stimulation of the enzyme. The oxidized (ferric) form is insensitive to NO and appears to be physiologically unimportant in cGMP signaling. In addition, a heme free state exists, which, like the oxidized state, does not elicit enzymatic activity via NO. 66 Identification of the 3 states of sGC has led to proposals that a redox shift occurs under pathological conditions. A shift in redox state from the ferrous to ferric state is associated with oxidative stress and production of ROS. This has been identified as one of several mechanisms in which the sGC/NO signaling pathway can be disrupted. Stasch et al 67 confirmed the presence of a sGC indistinguishable from the oxidized form of sGC found in vitro, in rat aorta, porcine pulmonary arterial endothelial cells, and human platelets.
Direct cGMP Targeting
Increasing intracellular concentration of cGMP has been shown to reduce myocyte contractility during reperfusion by inhibition of Na+/Ca2+ exchange and activation of SERCA via PKG-mediated phosphorylation of PLB. 68 3′,5′-Cyclic guanosine monophosphate-dependent protein kinase has also been shown to activate BKCa channels which when inhibited at reperfusion abolish protection afforded by upstream targets of cGMP. 69
D’Souza et al 70 reported that low concentrations of 8-bromo-cGMP (8Br-cGMP), a synthetic cGMP analog given just prior to ischemia through to early reperfusion limited infarct size in a rat isolated heart model. It was later shown that 8Br-cGMP also produced infarct limitation when given at reperfusion, reducing infarct size by 40% compared to control hearts. 71 These data demonstrated that targeting cGMP/PKG directly could afford infarct limitation mediated by elevating cGMP. 8-Bromo-cGMP has been reported to activate p38 mitogen-activated protein kinases in isolated adult rat cardiomyocytes but not extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), suggesting further components of the cytoprotective mechanism. 72
Soluble Guanylyl Cyclase Stimulating Compounds
The phenomenon of “nitrate tolerance” (tachyphylaxis to the vasodilator effects of organic NO donors) from the 1990s onward drove the discovery of compounds that can initiate the sGC/cGMP pathway independent of NO or when bioavailability of NO is low. Ko et al 73 described an indazole derivative, YC-1 (5-[1-(phenylmethyl)-1H-indazol-3-yl]-2-furanmethanol), later described as the first NO-independent, heme-dependent stimulator of sGC. 74 Subsequently, structurally related compounds were discovered that stimulate sGC, including 3-(4-amino-5-cyclopropylpyrimidine-2-yl)-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine (BAY 41-2272) and methyl N-[4,6-diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate (BAY 63-2521/Riociguat), see Table 1. 75 The mechanisms by which these compounds stimulate the activation of sGC are complex and go beyond the scope of this review. 73,75,76 However, their use has been investigated under pathological conditions where NO formation is compromised or when nitrate tolerance has developed. 77
List of Some of the Important Pharmacological Agents That Are Able to Positively Modify cGMP Concentrations.
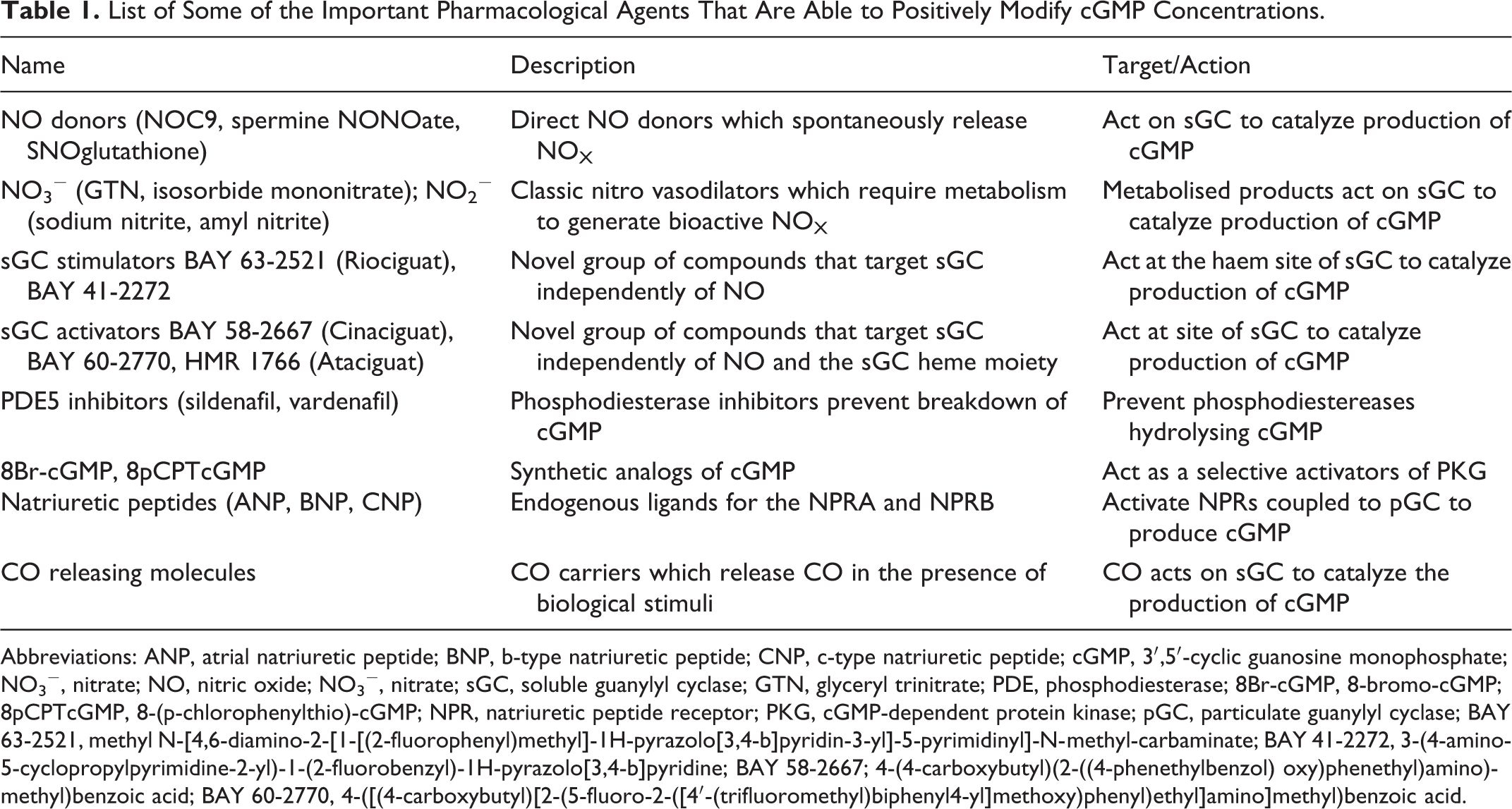
Abbreviations: ANP, atrial natriuretic peptide; BNP, b-type natriuretic peptide; CNP, c-type natriuretic peptide; cGMP, 3′,5′-cyclic guanosine monophosphate; NO3 −, nitrate; NO, nitric oxide; NO3 −, nitrate; sGC, soluble guanylyl cyclase; GTN, glyceryl trinitrate; PDE, phosphodiesterase; 8Br-cGMP, 8-bromo-cGMP; 8pCPTcGMP, 8-(p-chlorophenylthio)-cGMP; NPR, natriuretic peptide receptor; PKG, cGMP-dependent protein kinase; pGC, particulate guanylyl cyclase; BAY 63-2521, methyl N-[4,6-diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate; BAY 41-2272, 3-(4-amino-5-cyclopropylpyrimidine-2-yl)-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine; BAY 58-2667; 4-(4-carboxybutyl)(2-((4-phenethylbenzol) oxy)phenethyl)amino)methyl)benzoic acid; BAY 60-2770, 4-([(4-carboxybutyl)[2-(5-fluoro-2-([4′-(trifluoromethyl)biphenyl4-yl]methoxy)phenyl)ethyl]amino]methyl)benzoic acid.
Soluble Guanylyl Cyclase Activating Compounds
A second distinct class of sGC modulating compounds, described as sGC activators, was subsequently discovered. High throughput screening revealed several aminodicarboxylic acids including 4-(4-carboxybutyl)(2-((4-phenethylbenzol) oxy)phenethyl)amino)methyl)benzoic acid (BAY 58-2667). Described as the first and most potent NO-independent, heme-independent sGC activator, Stasch et al 78 demonstrated that BAY 58-2667 activated sGC even after it had been oxidized by the sGC inhibitor 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ) or even after losing its heme group completely. This class of compounds has become favored for use in the exploration of pathological diseases as they target the oxidized (pathological) state of the enzyme. In light of the proposed shift toward the oxidized and heme free states under conditions of oxidative stress, these compounds have been utilized in experimental models of ischemia–reperfusion where oxidant conditions are well documented. Investigation of the chemistry and pharmacology of these compounds demonstrates that sGC activation goes beyond NO binding to the heme site on the β subunit.
Specific sGC Targeting
Krieg et al 79 demonstrated significant infarct reduction in the rat heart when the sGC activator BAY 58-2667 was given at reperfusion following 30-minute global ischemia. They reported an elevation in cGMP levels in hearts perfused with the activator 40 times greater than in control hearts and further demonstrated that infusion of BAY 58-2667 in open chest in situ rabbits following left coronary artery occlusion limited infarct size by 40%. The protection was independent of NO but dependent on PKG and mitochondrial ATP-sensitive K channel signaling. In a similar model, Cohen et al 80 reported reduction in infarct size of 54%. Interestingly, the protection in that study was abrogated in the presence of a NOS inhibitor, an observation not concordant with the literature describing BAY 58-2667’s mechanism of action. 78 Salloum et al 81 have demonstrated that BAY 58-2667 can limit infarct size when administered pre- or postischemically in adult rabbits. They further suggested that PKG activity was increased following perfusion and interestingly H2S levels were elevated, abrogated by the PKG inhibitor KT5823. These data corroborate the most recent study, which demonstrates that BAY 58-2667 limited infarct size at reperfusion in mouse. This protective effect was abrogated when the same treatment protocol was utilized in cardiomyocyte specific PKG-I knockout mice. 82 Work from our own laboratory has demonstrated that perfusion of a structurally similar sGC activator 4-([(4-carboxybutyl)[2-(5-fluoro-2-([4′-(trifluoromethyl)biphenyl4-yl]methoxy)phenyl)ethyl]amino]methyl)benzoic acid (BAY 60-2770) could afford infarct limitation when perfused during reperfusion in an isolated rat heart model of regional ischemia. 83 This protection was not blocked by concomitant perfusion of ODQ, a heme site oxidizer. These data demonstrate that the ferric form of sGC is present in rabbit, rat, and mouse myocardium during ischemia, and early reperfusion as an elevation in cGMP was measured following perfusion of the sGC activator in this setting.
Heme Oxygenase and CO-Releasing Molecules
The microsomal enzyme heme oxygenase (HO) catalyzes the degradation of heme to CO, producing bilirubin and iron as coproducts. Heme oxygenase 1 is not constitutively present but can be activated under many pathological stimuli, including those present during ischemia–reperfusion such as oxidative stress and hypoxia. 84 Although CO-mediated activation of sGC only elicits small increases in catalytic activity, the antioxidant N-acetylcysteine and acute liver failure were able to limit infarct size in diabetic rats, which was abrogated by HO-1 inhibition. 85 3′,5′-Cyclic guanosine monophosphate levels were shown to be elevated in the aorta of spontaneously hypertensive rats following administration of the same HO-1 inducing agent, hemin. Induction of HO-1 by CO-releasing molecules has been shown to be cytoprotective when administered prior to ischemia–reperfusion. 86 The authors reported that the protection observed is comparable to that seen in classical late phase preconditioning; protection was conferred in part by recruiting HO-1. Taken together, these studies demonstrate that HO/ CO/sGC/cGMP signaling is impaired under pathological conditions in the myocardium, offering an additional target to afford cytoprotection (Figure 2).
Natriuretic Peptide Signaling
Natriuretic Peptides as an Indicator of Infarct Size
Natriuretic peptides play a fundamental role in cardiovascular homeostasis, their release being primarily regulated by pressure overload and myocardial stretch. In 1991, Mukoyama et al 87 reported for the first time that BNP levels were elevated during MI. It was later reported that elevation of BNP correlates with ischemia severity. 88,89 Since then several experimental studies have documented elevation of ANP 90 –92 and BNP 70,93 in models of myocardial ischemia. Most recently, numerous clinical studies confirmed the usefulness of measuring BNP levels in patients presenting with STEMI and suggest that they are good indicators of prognosis and infarct size. 94 –96
Natriuretic Peptides at Reperfusion
Several studies have identified that NPs administered at reperfusion afford protection in both rat and rabbit models of ischemia–reperfusion. Yang et al 97 reported that administration of ANP just prior to reperfusion limited infarct size in the rabbit heart. They also demonstrated that the protection afforded required KATP activity as inhibition of the channel with 5-hydroxydecanoate (5-HD) abrogated the protective effects of ANP. The B-type NP was also shown to limit infarct size in an in situ rat heart model, limiting infarct size in a concentration dependent manner. 98 Our laboratory reported similar results in an ex vivo rat heart model, demonstrating concentration-dependent infarct limitation with BNP, which like previous studies was dependent on KATP channel activity. 69 Furthermore, we reported that NPs could play a role in postconditioning-mediated protection as the nonspecific NPR-A/NPR-B antagonist isatin abolished the infarct limitation afforded by a 6 × 10-second postconditioning protocol in the rat heart. 69 In a cell viability assay, it has been reported that along with the sGC activator SNAP, BNP can limit cell death via common downstream signaling pathway involving elevated PKG. 99 Most recently, George et al 100 reported that perfusion of BNP for 7 days post-AMI significantly improved LV function and decreased LV remodeling in the rat heart. Atrial NP was administered to patients in the J-WIND clinical trial in which infarct size measured by creatine kinase was accessed. Reported outcomes of the trial suggested that patients receiving ANP had lower infarct size, fewer reperfusion injuries, and better outcomes than controls 101 (Table 2).
List of Clinical Trials (Completed and Ongoing) That Utilize Pharmacological Agents That Target NO/sGC/pGC/cGMP Signaling to Limit Infarct Size.

Abbreviations: AUC, area under the curve; J-WIND, Japan-Working Groups of Acute Myocardial Infarction for the Reduction of Necrotic Damage by KATP; hANP, human atrial natriuretic peptide; iv, intravenous; iNO, inducible nitric oxide; MI, myocardial infarction; MRI, magnetic resonance imaging.
Alternative NP Signaling Mediates Infarct Limitation
Infarct limitation afforded by NPs has been demonstrated to require KATP activity as described previously. It has also been demonstrated that protection is mediated by elevation of cGMP and distal PKG targets converging on the same effectors as NO/sGC signaling, that is, regulating calcium via PLB and L-type calcium channels and potassium efflux through the KATP channel. 41,70,102
Other signaling pathways have been suggested to play a part in NP-mediated infarct limitation. D’Souza et al
103
reported that concomitant perfusion of NG-nitro-
Similar observations were made in a reperfusion-targeted treatment by Ren et al
98
, who report that
Particulate Guanylyl Cyclase Independent Signaling
Historically regarded as a clearance receptor, NPR-C is devoid of guanylyl cyclase-coupled signaling, coupled to adenylyl cyclase inhibition through the inhibitory guanine nucleotide regulatory protein. Natriuretic peptide receptor C may in fact couple to signal transduction pathways in some cell types.
104,105
Interestingly, CNP signal transduction via NPR-C has been shown to be cardioprotective in the rat heart.
106
Surprisingly, this protection was potentiated in the presence of the NOS inhibitor
Most recently, a new class of so-called “designer” NPs is being developed. Cenderitide (CD) NP is the first in this class of NPs which coactivates both NPR-A and NPR-B. It has been shown to reduce LV mass in MI model rodents and cardiac unloading in dogs. 109 Initial clinical trials suggest a reduction in blood pressure in patients with stable heart failure as well as reduced creatine levels. It is proposed that continuous infusion of CD through a subcutaneous pump will improve patient outcome and reduce the duration of hospital stay. 110,111 Their cytoprotective benefit in models of myocardial ischemia–reperfusion is yet to be explored.
Phosphodiesterases
Phosphodiesterase Inhibition
Maintaining cGMP levels during ischemic insult by pharmacological manipulation of PDEs has been extensively investigated. The cGMP-specific PDE5 inhibitors have been utilized to explore this mechanism. A recent study has reported that as much as 20% of cGMP degradation in human myocytes is attributable to PDE5. 112 Expression levels of PDE5 are increased in the failing myocardium, and PDE5 inhibition has been shown to limit infarct size when administered prior to global ischemia in a mouse model. 113 The authors demonstrate that this protection is abolished when sildenafil was concomitantly perfused with the PKG blocker KT5823. They further concluded that the protection was associated with PKG-dependent phosphorylation of ERK, GSK3β, and increased expression of the prosurvival factor Bcl 2. 113 Early investigations demonstrated that sildenafil was cardioprotective in an in situ rabbit model, protection which was blocked by the KATP channel blocker 5-HD 114 and PKC inhibitor chelerythrine. 115 This has been corroborated using another PDE5 inhibitor vardenafil in combination with either the mitochondrial KATP channel blocker 5-HD or the sarcolemmal KATP channel blocker sodium salt of 1-[5-[2-(5-chloro-o-anisamido)ethyl]-2-methoxyphenyl]sulfonyl-3 methylthiourea (HMR 1098). 116 It was later demonstrated that sildenafil could induce delayed preconditioning via elevation of both iNOS and eNOS. Protection was blocked by the iNOS inhibitor 1400W. 117 Tadalafil, a PDE5 inhibitor with a longer half–life, was also able to limit infarct size in mouse 118,119 and rabbit. 120 More recently, protection has been demonstrated when sildenafil is perfused during early reperfusion. 121 The authors report that administration of the PDE5 inhibitor was associated with PLB Ser phosphorylation, also PKG dependent. 122 Interestingly, mouse hearts subjected to 30-minute global ischemia followed by 10-minute reperfusion with sildenafil did not elevate total cGMP beyond control level. This observation is corroborated by Elrod et al 123 who demonstrate that infarct limitation in a mouse model was independent of eNOS/NO/cGMP signaling and particularly that low level sildenafil (0.06 mg/kg) did not elevate myocardial cGMP levels. Further mechanistic studies have demonstrated that both sildenafil and vardenafil elicit their infarct limiting properties through a KATP channel-mediated mechanism 124 and that sildenafil improves left ventricular function in mice. 125
Regulation of Specific cGMP Pools
Spatially distinct pools of cGMP are now widely accepted; however, whether these discrete pools mediate individual and distinct actions remains to be fully demonstrated. Evidence supports the notion that PDEs which are localized by binding of GAF domains to proteins regulate specific cGMP pools, both in their cellular location but also via cAMP/PKA action through PDE2/3. Castro et al 19 reported that PKG activity can regulate cGMP concentrations in rat cardiac myocytes differentially via pGC and sGC. 3′,5′-Cyclic guanosine monophosphate-dependent protein kinase activated via pGC stimulates further cGMP production via positive feedback, the cGMP being localized to the sarcolemma via PDE2 control. Conversely, sGC-mediated PKG activity further enhances PDE5 activity limiting cGMP production and spatial distribution. They have previously demonstrated that cGMP-mediated activity varies depending on whether it is elevated via pGC or sGC, 126 further supporting the compartmentalization hypothesis. Most recently, Förster resonance energy transfer imaging in live neonatal rat cardiomyocytes expressing cGMP and cAMP markers has shown that sGC-mediated cGMP leads to activation of PKA RI via cAMP and a reduction in PKA RII. These opposing effects are mediated via PDE2/3 regulation, which are confined to specific cellular locations also. Interestingly, cAMP activity was not elevated via ANP/pGC signalling. 127 In light of cAMP/PKA in regulating calcium transients and inotropic effects, these observations may highlight further therapeutic targeting of specific cGMP/cAMP as a result of spatially distinct cGMP/PDE signaling.
Conclusions
The evidence suggests that sGC/cGMP signaling to limit infarct size is a tractable therapeutic target; however, it is clear that mass elevation of myocardial cGMP concentration in itself is not required to afford protection. Live cell imaging is now allowing real-time visualization of cGMP while exposing cells to pharmacological interventions. This information is required to ascertain how the dynamic signals mediate their action following differing stimuli, which cGMP elevating targets are important and which cGMP compartments may be key. With this information, suitable cytoprotective interventions to limit the injurious effects of ischemia–reperfusion injury via cGMP signaling could be developed.
Footnotes
Acknowledgment
The authors would like to acknowledge the continued support from The British Heart Foundation and Heart Research UK.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.