
Abstract
Postischemic accumulation of intracellular Na+ promotes calcium overload and contributes to cellular necrosis. Cardioprotection afforded by pharmacologic blockade of the sodium–hydrogen exchanger subtype 1 (NHE1) is thought to be more remarkable than that obtained by ischemic conditioning (IC). The window of protection provided by IC pretreatment is maintained even when performed up to 48 hours before ischemia. In addition, the perception exists that combined NHE1 inhibition plus IC produces greater than additive protection against ischemic injury. The current study compared the efficacy of NHE1 blockade by N-[2-methyl-4,5-bis(methylsulfonyl)-benzoyl]-guanidine (EMD 87580 5 mg/kg) combined with first- or second-window IC on ischemic tolerance in dogs subject to 90-minute acute ischemia and 180-minute reperfusion. Infarct size (tetrazolium staining), vascular responses, and myocardial perfusion (microspheres) were assessed. EMD 87580 given before ischemia or before reperfusion did not reduce infarct size (compared to vehicle-treated group). Significant protection against tissue necrosis was obtained by both first- and second-window IC, but additive cardioprotection (ie, greater than that afforded by IC) was not observed by treatment with EMD 87580. Vascular reactivity in the infarct-related artery was not preserved after ischemia–reperfusion in any of the experimental groups. Likewise, either the pharmacologic or the nonpharmacologic interventions did not modify myocardial perfusion. These data demonstrate that EMD 87580 did not protect against ischemia–reperfusion injury regardless of the time of drug administration. Combined EMD 87580 and IC did not antagonize protection that was achieved by either first- or second-window IC alone; no additive protection beyond preconditioning was obtained. Further study is necessary to assess the value of NHE1 blockers as protective agents against myocardial injury.
Introduction
Robust protection against ischemic injury afforded by sodium–hydrogen exchanger subtype 1 (NHE1) blockers in animal studies has heightened expectations for successful translation to patients. Rupprecht and coworkers documented that intravenous cariporide improved myocardial function in patients with acute myocardial infarction treated by percutaneous transluminal coronary angioplasty 1 ; however, success of different clinical studies [ie, evaluation of the safety and cardioprotective effects of eniporide on acute myocardial infarction (ESCAMI), GUARD during ischemia against necrosis (GUARDIAN), sodium–hydrogen Exchange inhibition to Prevent coronary Events in acute cardiac condition (EXPEDITION)] is mitigated. 2 In the ESCAMI (phases 1 and 2) trial, eniporide administered prior to reperfusion did not reduce infarct size or improve clinical outcomes. 3 Similarly, the GUARD During Ischemia Against Necrosis (GUARDIAN) trial did not provide significant protection against infarction or reduce primary end points; but some benefit was observed in high-risk coronary artery bypass patients. 4,5 The sodium-hydrogen Exchange inhibition to Prevent coronary Events in acute cardiac condition (EXPEDITION) trial examined the effects of cariporide on nonfatal myocardial infarction; however, mortality risk was elevated due to an increased number of cerebrovascular events. 2,6
Ischemic conditioning (IC; pre, per, post, remote, anesthetic, pharmacologic, and so on) affords significant protection against most experimental and clinical end points after acute ischemic injury. The IC-mediated protection occurs in 2 phases: the first window develops within minutes of the initial ischemic event and lasts 2 to 3 hours, 7 while the second window, or late phase, occurs between 24 and 72 hours. 8,9 Mechanisms responsible for each phase of protection are remarkably diverse and may even be shared (cf 10 –13 for reviews). The ability of IC, independent of time of application, to delay the development of tissue injury is reduced in relation to the duration of ischemia 14 ; enhancement of the so-called ceiling of protection is potentially achievable by combining pharmacologic with preconditioning treatments. 15
Numerous endogenous mechanisms are recruited during IC that stimulate the release of trigger compounds (ie, prosurvival kinases) that activate intracellular signaling pathways and lead to increased phosphorylation of target proteins. 16 The NHE1 (predominant myocardial isoform for the proton extrusion pathway 17 ) may be important in IC-mediated protection; additionally, IC and NHE1 blockade may share a common mechanism. 18 Gumina and coworkers tested the hypothesis that NHE1 inhibition could be more effective than IC in attenuating myocardial infarction and showed that first-window IC and NHE1 blockade with BIIB 513 (benzamide-N-(aminoiminomethyl)-4-[4-(2-furanylcarbonyl)-1-piperazinyl]-3-(methylsulfonyl) methanesulfonate) produced greater-than-additive protection in canines. 15 In addition, they reported that NHE1 blockade prior to acute coronary occlusion conferred greater cardioprotection than IC alone. Whether NHE1 blockade confers greater myocardial protection than second-window IC protection has not been studied.
The purpose of this study was to investigate the cardioprotective efficacy of N-[2-methyl-4,5-bis(methylsulfonyl)-benzoyl]-guanidine (EMD) 87580, a selective inhibitor of the NHE1 isoform, administered before acute ischemia or before reperfusion, against myocardial and vascular injury in an in vivo canine preparation. An additional goal was to compare efficacy of either first- or second-window (48 hours before acute ischemia/reperfusion) IC pretreatment combined with NHE1 blockade on the development of myocardial injury.
Materials and Methods
Adult mongrel dogs were treated in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication 85-23, revised 1996); experimental procedures were performed in accordance with the “Guide to the Care and Use of Experimental Animals” (vols. 1 and 2) of the Canadian Council on Animal Care. Laval University is compliant with these guidelines (A5012-01); the Laval University Animal Ethics Committee approved these studies. All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Dogs were acquired through the Division of Laboratory Animal Services at Laval University; they were housed in individual cages under conditions of constant temperature and humidity and kept on a strict 12:12-hour dark–light cycle. Dogs had free access to food and water.
Materials
In these studies, we used the selective NHE1 isoform inhibitor, EMD 87580 (N-[2-methyl-4,5-bis-(methylsulfonyl)-benzoyl]-guanidine), a benzoylguanidine derivative of amiloride. Drug was provided by Merck KGaA, Darmstadt, Germany; this compound is the hydrochloride salt that crystallizes as a monohydrate; is stable in neutral, acidic, and alkaline solutions; and is soluble in physiologic saline.
General Surgical Preparation
Mongrel dogs of either sex, weighing 20 to 25 kg, were premedicated with diazepam (1 mg/kg intravenously [IV]) and fentanyl (20 μg/kg IV) and then anesthetized using sodium pentobarbital (25 mg/kg IV). Dogs were intubated and mechanically ventilated with room air supplemented with 100% oxygen. Atelectasis was prevented by maintaining an end-expiratory pressure of 5 to 7 cm H2O with a trap. Arterial blood pH, PO2, and PCO2 was monitored regularly and maintained within normal physiological limits. Body temperature was kept at 38°C ± 1°C by a water-jacketed Micro-Temp heating unit (Zimmer, Dover, Ohio, USA), since temperature-induced variability during ischemia/reperfusion is a recognized determinant of myocyte necrosis 19 ; temperature was measured continuously with probes positioned in the abdomen (just below the diaphragm) and trachea (in the endotracheal tube).
In the supine position, vascular introducer sheaths (8F; Terumo Medical Corp, Mississauga, ON, Canada) were positioned in the left and right femoral arteries: A triple-lumen central venous catheter (7F; Arrow-Howes, Arrow Intl Inc, Reading, Pennsylvania) was placed in the right femoral vein for administration of drugs and fluids. After splenectomy through a mid-line abdominal incision, 20 a left thoracotomy was performed and the heart suspended in a pericardial cradle; catheters were placed in the internal thoracic artery (for withdrawal of reference blood for calculation of regional blood flow with microspheres), coronary sinus, and left atrium. A 5F micro-tipped pressure transducer (MPC500; Millar Instruments Inc, Houston, Texas, USA) was placed in the left ventricle (LV) cavity through the apex to measure LV pressure (LV dP/dt was recorded by electronic differentiation of the LV pressure signal); a 7F pigtail catheter was advanced to the aortic root via the left femoral artery to measure arterial pressure. The left circumflex coronary artery proximal to the first marginal branch was dissected free and a snare positioned. A catheter was inserted in the lumen of this artery immediately distal to the snare occluder to measure coronary artery pressure. 21 Phasic coronary blood flow was measured with a transit time flowmeter (T206; Transonic Systems Inc, Ithaca, New York); the chest cavity was covered with plastic film to prevent undue myocardial cooling.
Dogs were randomly assigned to either of the 3 study protocols shown in Figure 1; a total of 9 experimental groups were studied.

Schema of experimental protocols. Dogs were exposed to 90-minute acute ischemia (IS) and 180-minute reperfusion (REP); in Protocol A (no IC), a 40-minute wait period was allowed to compensate for time required for IC (4 cycles of 5-minute IS/5-minute REP). Reactive hyperemia (RH) was assessed before IS and during reperfusion (30 and 180 minutes). Saline (VEH) or EMD 87580 was administered intravenously (IV) either before IS or before REP. IC indicates ischemic conditioning.
Protocol A: EMD 87580
After surgical preparation, dogs (n = 8/group) were assigned to the following groups: 1—sham [vehicle (VEH), saline IV during ischemia–reperfusion], 2—EMD 87580 (5 mg/kg bolus over 5-minute, IV) before onset of acute regional ischemia (EMDI), and 3—EMD 87850 (5 mg/kg bolus over 5-minute, IV) before onset of reperfusion (EMDR).
A reactive hyperemic (RH) response of the left circumflex artery was recorded under steady-state hemodynamic conditions prior to coronary occlusion 22 ; vascular reserve was assessed by 20-second occlusion of the infarct-related artery. A 40-minute wait period was instituted to allow comparisons with IC studies. Subsequently, dogs underwent 90-minute regional ischemia followed by 180-minute reperfusion; all dogs were given a 10 mg IV bolus of xylocaine (Astra Pharma, Inc, Mississauga, Ontario, Canada) at 30-minute ischemia and prior to reperfusion to limit ischemia- or reperfusion-induced dysrhythmias. During reperfusion, RH in the infarct-related coronary vessel was measured at 30 and 175 minutes. Microspheres were injected into the left atrium at (1) baseline, (2) 30-minute ischemia, (3) 30-minute reperfusion, and (4) 180-minute reperfusion. Hearts that fibrillated were cardioverted (direct current [DC] shock ≤50 J) with a cardiac defibrillator (general electric); if defibrillation was not successful after 2 attempts, the animal was euthanized and not entered into the data analysis. Cardiac dysrhythmias (tachycardia and fibrillation) were considered terminated if they were followed by at least 3 or more consecutive normal sinus beats.
Protocol B: First-Window IC + EMD 87580
Dogs (n = 8/group) were assigned to 1 of the 3 treatments as described in Protocol A. Coronary vascular reserve was evaluated by reactive hyperemia as described earlier. After return to steady state, dogs underwent classical first-window IC (4 cycles of 5-minute ischemia and 5-minute coronary reperfusion). 7 Subsequently, dogs underwent 90-minute ischemia and 180-minute reperfusion; reactive hyperemia in the infarct-related coronary vessel was evaluated at 30- and 175-minute reperfusion. Microspheres were injected into the left atrium at (1) baseline, (2) 30-minute ischemia, (3) 30-minute reperfusion, and (4) 180-minute reperfusion. EMD 87580 was administered as described earlier, and all dogs were given xylocaine (10 mg IV; Astra Pharma, Inc) after 30-minute ischemia and prior to reperfusion. The exclusion criteria described earlier were used in the event of unsuccessful cardioversion.
Protocol C: Second-window IC + EMD 87580
Dogs (n = 8/group) were premedicated with diazepam (1 mg/kg IV) and fentanyl (20 μg/kg IV) and anesthetized with 1.5% to 2.0% isoflurane. The IC was performed by coronary catheterization as described previously 23 ; briefly, the right femoral artery was cannulated with a 7F sheath for percutaneous transluminal coronary angioplasty, and 1000 IU heparin was administered IV. Under angiographic guidance, a 6F guiding catheter was inserted and advanced to the coronary ostium. Left coronary angiography was performed in oblique view to delineate the left main coronary artery and its branches; a floppy guidewire (0.0014 inch) was advanced into the left main circumflex artery, and an inflatable balloon catheter was positioned just distal to the first marginal branch defined by angiography. Second-window IC was performed by balloon inflation (3 bars)/deflation (4 cycles of 5-minute ischemia and 5-minute reperfusion); 1 minute after either onset of occlusion or reperfusion, contrast medium was injected and an angiogram obtained to verify the absence or restoration of blood flow within the infarct-related artery. After completion of the IC protocol, catheters were removed and tissue and skin incisions sutured; dogs were weaned from the respirator and extubated after restoration of a regular breathing pattern. Each dog was administered antibiotic (cefazoline sodium, 5.5 mg/kg, intramuscular [im]), analgesic (buprenorphine, 0.05 mg/kg, im), and heparin (500 IU, subcutaneously) before returning to the recovery room.
After 48-hour recovery, dogs were assigned to 1 of the 3 treatments described in Protocol A. Dogs underwent the surgical and instrumentation protocols; coronary vascular reserve was evaluated, and then, dogs underwent 90-minute ischemia (same position as determined for the IC procedure) followed by 180-minute reperfusion. Reactive hyperemia in the infarct-related coronary vessel was measured at 30- and 175-minute reperfusion. Microspheres were injected into the left atrium at (1) baseline, (2) 30-minute ischemia, (3) 30-minute reperfusion, and (4) 180-minute reperfusion. Xylocaine (10 mg IV bolus; Astra Pharma, Inc) was given at 30-minute ischemia and before reperfusion; exclusion criteria in the event of unsuccessful cardioversion were the same as above.
Plasma Concentrations of EMD 87580
Venous blood (femoral vein) was obtained before IV administration (ie, 0 minute) of EMD 87580, 5 and 60 minutes after IV administration, and at the end of the experiment. Plasma levels of EMD 87580 were determined using high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) as described by Smilde and coworkers 24 and were performed in a blinded analysis at Merck KGaA. Characteristics of EMD 87580, in terms of potency and selectivity compared to other NHE isoforms, have been reported. 25,26
Hemodynamic Measurements
Standard lead II of the scalar electrocardiogram (ECG), aortic and LV pressures, coronary sinus, coronary artery pressure, and phasic coronary blood flow (infarct-related artery) were recorded continuously on a 12-channel direct-writing oscillograph (Yokogawa OR1200A; Electro-Meters, Dorval, Quebec, Canada) and on videotape using a TEAC XR-510 data recorder for later analysis.
Infarct Size, Myocardial Blood Flow, and Coronary Vascular Reserve Analysis
Regional myocardial blood flow was determined using microspheres suspended in saline (15 μm; NEN, Boston, Massachusetts). 27 At the end of each study, the left circumflex coronary artery was ligated at the site of previous occlusion. Under deep pentobarbital anesthesia, cardiac arrest was induced with saturated potassium chloride; 10 mL of Monastral blue dye (3%, w/v) was injected intra-atrially to delineate area at risk as previously described, 28 and afterward, warmed (37°C) 2,3,5-triphenyltetrazolium chloride (1.5%) was infused into the ischemic region for 30 minutes. The heart was excised, rinsed in saline, and the atria, large epicardial vessels, and right ventricle removed; the heart was fixed in 10% buffered formaldehyde. The LV was sliced (5-mm thick) from the apex to base, and the outline of each slice [ie, total LV area (LVA)], necrotic area (AN), and the anatomic area at risk (AAR) were traced onto acetates for determination of infarct size; predetermined exclusion criteria for AAR (≤20% of LVA) were used to ensure anatomic consistency. The LVA, AAR, and AN were measured using a digitizing tablet (Summagraphics II Plus) interfaced with a personal computer and analyzed with Sigma Scan software (SPSS Inc, California). From the same slices, distribution of ventricular blood flow was assessed in myocardium obtained from the central portion of the nonischemic and ischemic zones; these were subdivided into endocardial, mid-myocardial, and epicardial segments. Tissue samples were weighed, and gamma activity in biopsies and blood reference samples were measured in a gamma-well scintillation counter (Cobra II, Canberra Packard Instruments, Nepean, ON, Canada) with standard window settings as described previously. 27 Tissue counts were corrected for background, decay, and isotope spillover; regional myocardial blood flow (expressed as mL/min/g of myocardium) was calculated with computer software (PCGERDA, Packard Instruments).
Reactive hyperemia responses were measured from strip chart recordings; baseline (Qbase) and peak (Qpeak) flow were determined as described previously. 29,30 Coronary vascular reserve was determined as the quotient of Qpeak/Qbase. Coronary vascular conductance (CVC) at baseline and at peak flow during reactive hyperemia was calculated as the quotient of diastolic coronary blood flow and diastolic coronary perfusion pressure.
Data Analysis
Differences in cardiac hemodynamics, phasic coronary blood flow, and myocardial blood flow were determined by analysis of variance (ANOVA), and multiple comparisons were performed using the Student-Newman-Keuls multiple range test. The relation between infarct size and myocardial blood flow was analyzed using a multivariate analysis; infarct size (normalized to risk area) was the dependent variable, and blood flow within the inner two-thirds of the ischemic zone was the covariate. A probability (P) level of ≤.05 was considered statistically significant; normality and variance assumptions were fulfilled. The influence of ventricular tachycardia/fibrillation on survival after the ischemia–reperfusion insult was determined using the Fischer exact test and chi-square analysis; all statistical analyses were done using SAS software (SAS Institute Inc, Cary, North Carolina). Sample size determination for these studies was based on the provision of a 90% power to detect, at a P ≤ .05 significance level, a minimum 20% reduction/augmentation (expected standard deviation of 8%) in infarct size.
Results
Seventy-five dogs were entered into the study; 1 was excluded due to technical difficulties during surgery. Two dogs (1 from VEH treated—Protocol A; 1 from EMDI—Protocol C) could not be resuscitated after 2 attempts at cardioversion and succumbed to ventricular fibrillation; only 2 attempts of defibrillation (with paddles directly on the heart) were made, since significant modulation of different parameters including vascular reserve, blood flow, and cardiac function are apparent with repetitive defibrillation attempts (unpublished data). Thus, 72 dogs completed the protocol and were used in the data analysis.
Plasma EMD 87580 Levels
Optimal dose and mode of delivery of EMD 87580 to ischemic myocardium may be critical for efficacy of NHE1 inhibitors. Plasma EMD 87580 levels were verified as shown in Figure 2. Five minutes after bolus intravenous injection, plasma levels reached their highest level; plasma concentration decrease was initially rapid (t½ ∼ 55-minute) with an apparent elimination half-life (t½) ∼3.8 hours over the remainder of the experiment—the latter value corresponds to pharmacokinetic findings reported for humans given EMD 87580 orally. 24 Plasma levels were within recommended levels for the duration of the ischemia–reperfusion protocols. 24,31

Plasma levels of EMD 87580 versus time; data are means ± 1 SD. The chemical structure of EMD 87580 (N-[2-methyl-4,5-bis(methylsulfonyl)-benzoyl]-guanidine) is shown in the inset.
Protocol A: EMD 87580
Cardiac hemodynamics
Hemodynamic data for the 3 treatment groups (VEH, EMDI, and EMDR) are summarized in Table 1; acute ischemia resulted in lower LV pressure in VEH and EMDI groups; in EMDR dogs, no change was observed for this parameter. As expected, pressure and flow in the infarct-related artery were substantially lower during coronary occlusion. Rate pressure product (RPP), an index used to represent myocardial oxygen demand, was slightly lower during reperfusion compared to baseline in EMDI dogs and was more pronounced in EMDR dogs.
Summary of Cardiac Hemodynamic Data With No IC.

Abbreviations: IS, ischemia; REP, reperfusion; VEH, dogs given saline; EMDI, EMD 87580 prior to IS; EMDR, EMD 87580 before REP; HR, heart rate (beats/min); LVPs (mm Hg), systolic LV pressure; LVPd (mm Hg), diastolic LV pressure; CPs (mm Hg), systolic coronary artery pressure; CPd (mm Hg), diastolic coronary artery pressure; CSs (mm Hg), systolic coronary sinus pressure; CSd (mm Hg), diastolic coronary sinus pressure; RPP (beats/min × mm Hg/1000), rate pressure product; QCor (mL/min), phasic coronary artery blood flow.
aData are mean ± 1 SD. Multiple comparisons between experimental groups and within interventions were done by ANOVA with the Student-Newman-Keuls multiple range test.
bP ≤ .05 vs baseline value for each group.
Changes in coronary reactive hyperemia caused by ischemia–reperfusion injury are summarized in Table 2. Qbase decreased overall (compared to baseline values) by the end of reperfusion (REP180); Qpeak was significantly lower in all experimental groups which suggests an overall loss of coronary flow reserve (Qpeak/Qbase), despite treatment with EMD 87580. Similar changes in CVC (CVCbase, CVCpeak) were also observed.
Changes in Coronary Reserve of Infarct-Related Artery.a
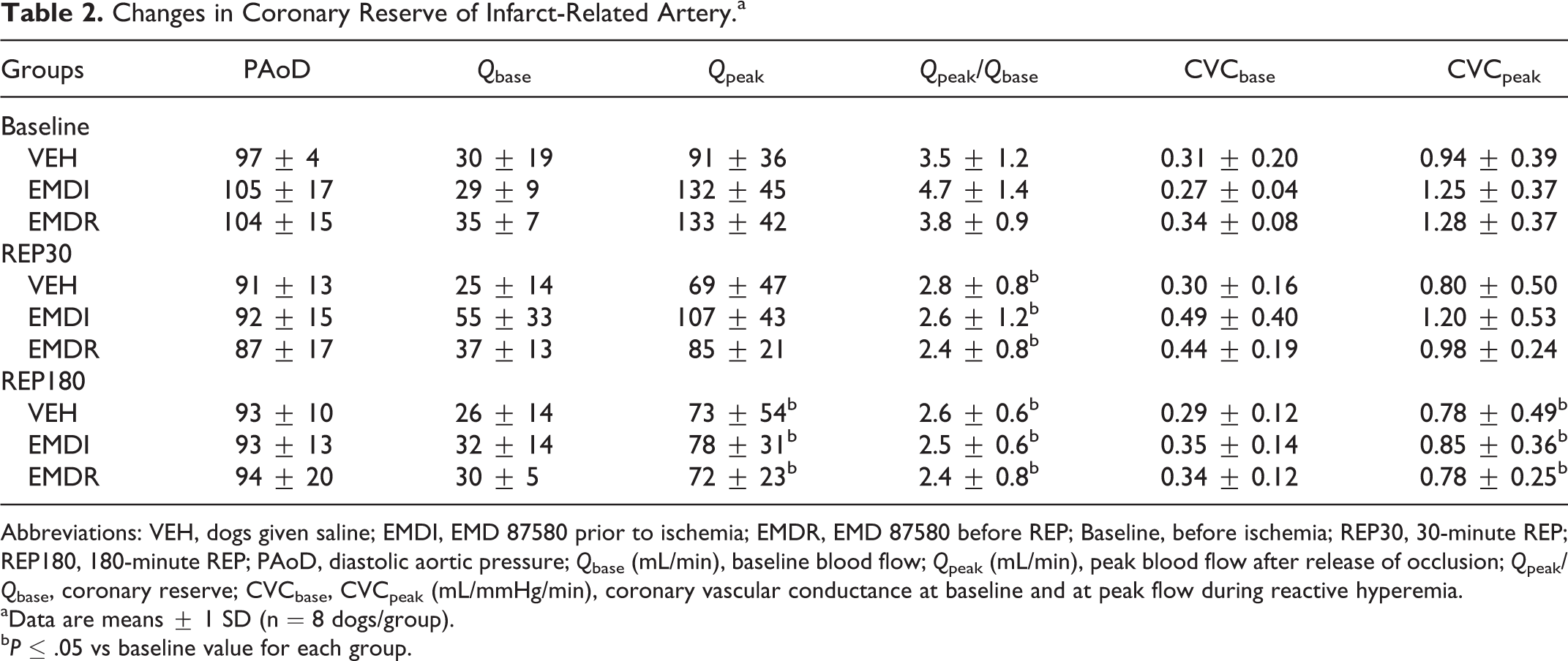
Abbreviations: VEH, dogs given saline; EMDI, EMD 87580 prior to ischemia; EMDR, EMD 87580 before REP; Baseline, before ischemia; REP30, 30-minute REP; REP180, 180-minute REP; PAoD, diastolic aortic pressure; Qbase (mL/min), baseline blood flow; Qpeak (mL/min), peak blood flow after release of occlusion; Qpeak/Qbase, coronary reserve; CVCbase, CVCpeak (mL/mmHg/min), coronary vascular conductance at baseline and at peak flow during reactive hyperemia.
aData are means ± 1 SD (n = 8 dogs/group).
bP ≤ .05 vs baseline value for each group.
Regional transmural myocardial blood flow in all dogs given either VEH or EMD 87580 before ischemia or reperfusion followed a similar profile (cf. Table 3); REP180 myocardial perfusion was restored to near baseline values in all groups. Compensatory adjustments in myocardial perfusion of the adjacent nonischemic myocardium were not observed during the 90-minute ischemic period.
Myocardial Perfusion in Ischemic and Nonischemic Regions.a
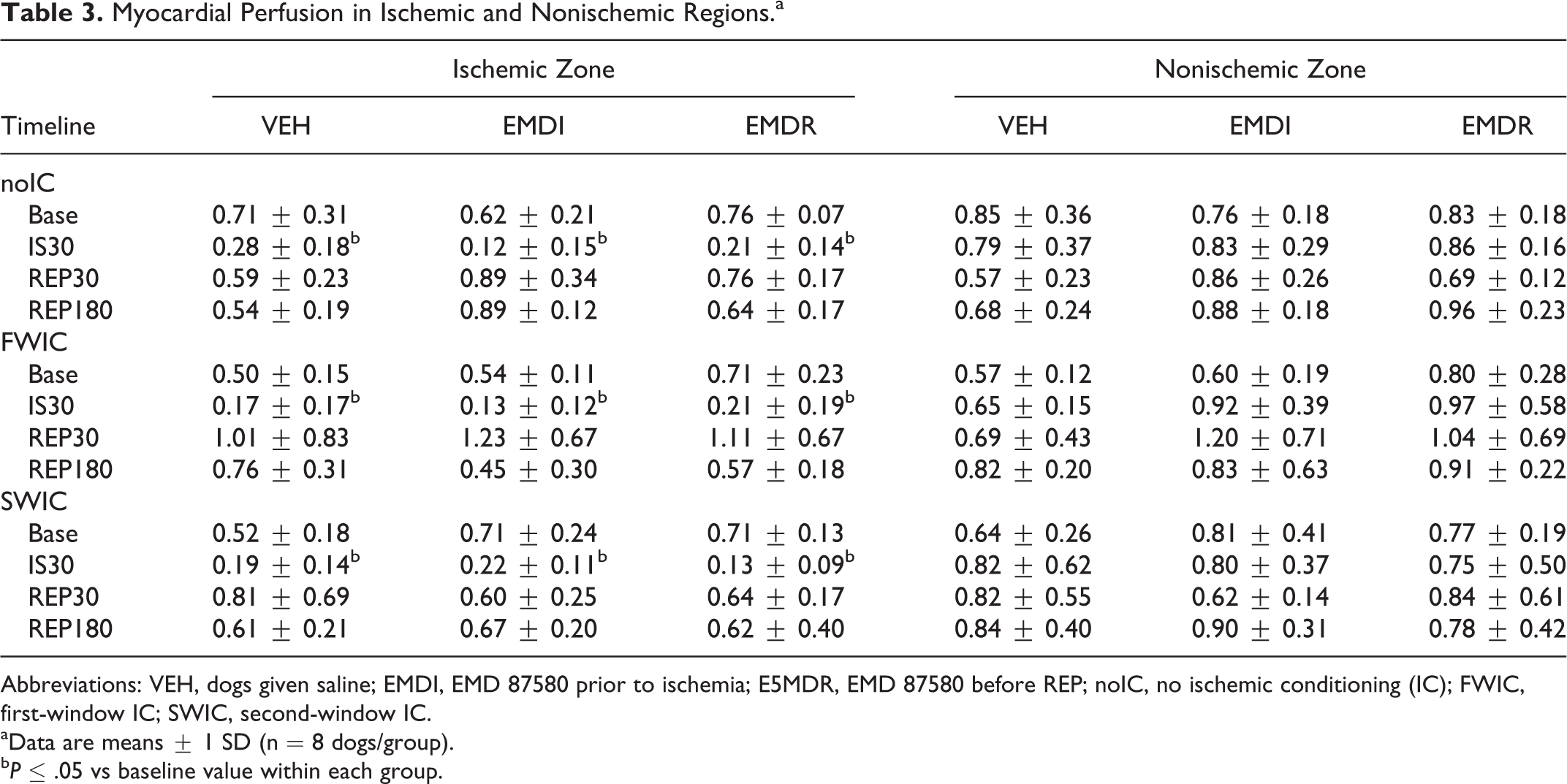
Abbreviations: VEH, dogs given saline; EMDI, EMD 87580 prior to ischemia; E5MDR, EMD 87580 before REP; noIC, no ischemic conditioning (IC); FWIC, first-window IC; SWIC, second-window IC.
aData are means ± 1 SD (n = 8 dogs/group).
bP ≤ .05 vs baseline value within each group.
The AAR (as percent LVA) was similar for all of the experimental groups (VEH, 47.8 ± 7.2; EMDI, 48.3 ± 4.5; EMDR, 46.1 ± 4.9; P = .556, ANOVA) as shown in Figure 3. Infarct size (as percent AAR) was not significantly smaller after EMD 87580 given before ischemia or reperfusion (VEH, 31.6 ± 10.4; EMDI, 28.8 ± 11.9; EMDR, 24.4 ± 9.9; P = .429, ANOVA). The degree of ischemia was similar between groups as shown by coronary collateral blood flow (mL/min/g) levels within the endocardial layer of the AAR (VEH, 0.09 ± 0.05; EMDI, 0.06 ± 0.03; EMDR, 0.09 ± 0.04; P = .225, ANOVA); blood flow (mL/min/g) in adjacent nonischemic myocardium measured at the same time point was VEH, 0.79 ± 0.37; EMDI, 0.83 ± 0.29; EMDR, 0.86 ± 0.16 (P = .171, ANOVA). Canine myocardium is known to have a higher level of coronary collateral circulation that could affect the development of tissue necrosis; accordingly, the infarct size/coronary collateral blood flow relation was analyzed for each experimental group. In VEH-treated dogs, the expected inverse relation between coronary collateral flow and infarct size was obtained (low flow correlates to larger infarcts); this relation was not shifted with EMD 87580 treatment which suggests that the drug is unable to protect against ischemic injury.

Risk zone size (as % LV area) and ANAR (as % AAR for dogs that were not pretreated by ischemic conditioning). Results are means ± 1 SD (each data point represents an individual dog). The inverse relation between myocardial infarct size and endocardial blood flow in the area at risk is shown (along with 95% confidence limits) for VEH (filled circles; y = 36.4-83.66x, r2 = .286). Results for EMDI (open squares) and EMDR (open triangles) treatment groups did not produce a downward shift of the relation. LV area indicates left ventricle area; AAR, anatomic area at risk.
Protocol B: First-Window IC + EMD 87580
Cardiac hemodynamics
Hemodynamic data for these studies are summarized in Table 4; acute ischemia did not affect LV pressure (systolic/diastolic) during these experiments. As expected, pressure and flow in the infarct-related artery were substantially lower during coronary occlusion. The RPP was similar to baseline values after 180-minute reperfusion; these findings were not affected by treatment with EMD 87580.
Summary of Cardiac Hemodynamic Data in Dogs With First-Window IC.

Abbreviations: IS, ischemia; REP, reperfusion; VEH, dogs given saline; EMDI, EMD 87580 prior to IS; EMDR, EMD 87580 before REP; HR, heart rate (beats/min); LVPs (mm Hg), systolic LV pressure; LVPd (mm Hg), diastolic LV pressure; CPs (mm Hg), systolic coronary artery pressure; CPd (mm Hg), diastolic coronary artery pressure; CSs (mm Hg), systolic coronary sinus pressure; CSd (mm Hg), diastolic coronary sinus pressure; RPP (beats/min × mm Hg/1000), rate pressure product; QCor (mL/min), phasic coronary artery blood flow.
aData are mean ± 1 SD. Multiple comparisons between experimental groups and within interventions were done by ANOVA with the Student-Newman-Keuls multiple range test.
bP ≤ .05 vs baseline value for each group.
Changes in coronary reactive hyperemia are summarized in Table 5. Qbase remained near baseline levels by the end of reperfusion (REP180); Qpeak at REP180 was significantly lower in all experimental groups indicating an overall loss of coronary flow reserve, despite treatment with EMD 87580. Similar changes in CVC were also observed for these groups.
Change in Vascular Reserve of Infarct-Related Artery During Reactive Hyperemia in Dogs Subject to First-Window IC.a

Abbreviations: VEH, dogs given saline; EMDI, EMD 87580 prior to ischemia; EMDR, EMD 87580 before REP; Baseline, before ischemia; REP30, 30-miuten REP; REP180, 180-minute REP; PAoD, diastolic aortic pressure; Qbase (mL/min), baseline blood flow; Qpeak (mL/min), peak blood flow after release of occlusion; Qpeak/Qbase, coronary reserve; CVCbase, CVCpeak (mL/mmHg/min), coronary vascular conductance at baseline and at peak flow during reactive hyperemia.
aData are means ± 1 SD (n = 8 dogs/group).
bP ≤ .05 vs baseline value for each group.
The AAR (as percent LVA) was similar for all of the experimental groups (VEH, 43.6 ± 7.8; EMDI, 42.1 ± 6.9; EMDR, 43.4 ± 9.9; P = .501, ANOVA) as shown in Figure 4. Infarct size (as percent AAR) was not significantly smaller after EMD 87580 given before ischemia or reperfusion (VEH, 17.4 ± 5.8; EMDI, 19.6 ± 8.3; EMDR, 18.4 ± 7.4; P = .152, ANOVA). The extent of ischemia was similar between groups as shown by coronary collateral blood flow (mL/min/g) levels within the endocardial layer of the AAR (VEH, 0.08 ± 0.06; EMDI, 0.06 ± 0.03; EMDR, 0.10 ± 0.04; P = .222, ANOVA); blood flow (mL/min/g) in adjacent nonischemic myocardium measured at the same time point was VEH, 0.65 ± 0.15; EMDI, 0.92 ± 0.39; and EMDR, 0.97 ± 0.58 (P = .438, ANOVA). Since infarct size is influenced by the level of coronary collateral perfusion of the risk region in canines, the relation between these variables was examined for each treatment group; in the VEH group, an inverse relation was obtained (low flow correlates to larger infarcts). The relationship was not shifted by the treatment with EMD 87580.

Risk zone size (as % LV area) and ANAR (as % AAR) for VEH (open bar), EMDI, and EMDR (hatched bars) for dogs pretreated by first-window ischemic conditioning. Results are means ± 1 SD (each data point represents an individual dog). The relation between myocardial infarct size and endocardial blood flow in the area at risk (along with 95% confidence limits) is illustrated. Results for VEH (filled circles; y = 18.06-8.35x, r2 = .008), EMDI (open squares), and EMDR (open triangles) groups were similar in agreement with the consistent level of cardioprotection observed in these dogs. LV area indicates left ventricle area; AAR, anatomic area at risk.
Protocol C: Second-Window IC + EMD 87580
Cardiac hemodynamics
Hemodynamic data for the 3 treatment groups (VEH, EMDI, and EMDR) are summarized in Table 6; acute coronary occlusion produced a small but nonsignificant lowering of LV pressure but was not different from baseline levels at the end of reperfusion. As expected, pressure and flow in the infarct-related artery were substantially lower during coronary occlusion. RPP was similar to baseline values after 180-minute reperfusion; these findings were not affected by treatment with EMD 87580.
Summary of Cardiac Hemodynamic Data in Dogs With Second-Window IC.a

Abbreviations: IS, ischemia; REP, reperfusion; VEH, dogs given saline; EMDI, EMD 87580 prior to IS; EMDR, EMD 87580 before REP; HR, heart rate (beats/min); LVPs (mm Hg), systolic LV pressure; LVPd (mm Hg), diastolic LV pressure; CPs (mm Hg), systolic coronary artery pressure; CPd (mm Hg), diastolic coronary artery pressure; CSs (mm Hg), systolic coronary sinus pressure; CSd (mm Hg), diastolic coronary sinus pressure; RPP (beats/min × mm Hg/1000), rate pressure product; QCor (mL/min), phasic coronary artery blood flow.
aData are mean ± 1 SD. Multiple comparisons between experimental groups and within interventions were done by ANOVA with the Student-Newman-Keuls multiple range test.
bP ≤ .05 vs baseline value for each group.
Changes in coronary coronary flow reserve caused by ischemia–reperfusion injury are summarized in Table 7. Qbase was not significantly changed by REP180; however, Qpeak was significantly lower in all experimental groups indicating the overall loss of coronary flow reserve, despite treatment with EMD 87580. Similar changes in CVC were also observed for these groups.
Change in Vascular Reserve of Infarct-Related Artery During Reactive Hyperemia in Dogs Subject to Second-Window IC.a

Abbreviations: VEH, dogs given saline; EMDI, EMD 87580 prior to IS; EMDR, EMD 87580 before REP; Baseline, before ischemia; REP30, 30-minute REP; REP180, 180-minute REP; PAoD, diastolic aortic pressure; Qbase (mL/min), baseline blood flow; Qpeak (mL/min), peak blood flow after release of occlusion; Qpeak/Qbase, coronary reserve; CVCbase, CVCpeak (mL/mm Hg/min), coronary vascular conductance at baseline and at peak flow during reactive hyperemia.
aData are means ± 1 SD (n = 8 dogs/group).
bP ≤ .05 vs baseline value for each group.
The AAR (as percent LVA) was similar for all of the experimental groups (VEH, 40.5 ± 6.0; EMDI, 39.3 ± 4.2, EMDR, 40.3 ± 6.1; P = .362, ANOVA) as shown in Figure 5. Infarct size (as percent AAR) was not significantly smaller after EMD 87580 given before ischemia or reperfusion (VEH, 17.4 ± 5.7; EMDI, 13.3 ± 6.8, EMDR, 16.7 ± 8.4; P = .156, ANOVA). The extent of ischemia was similar between groups as shown by coronary collateral blood flow (mL/min/g) levels within the endocardial layer of the AAR (VEH, 0.09 ± 0.06; EMDI, 0.13 ± 0.02, EMDR, 0.10 ± 0.05; P = .641, ANOVA); blood flow (mL/min/g) in adjacent nonischemic myocardium measured at the same time point was VEH, 0.82 ± 0.62; EMDI, 0.80 ± 0.37; and EMDR, 0.75 ± 0.50 (P = .118, ANOVA). An inverse relation between coronary collateral flow and infarct size that was obtained for VEH-treated dogs was not shifted with EMD 87580 treatment.

Risk zone size (as % LV area) and ANAR (as % AAR) for VEH (open bar), EMDI, and EMDR (hatched bars) for dogs pretreated by second-window ischemic conditioning. Results are means ± 1 SD (each data point represents an individual dog). The relation between myocardial infarct size and endocardial blood flow in the area at risk (along with 95% confidence limits) is illustrated. Results for VEH (filled circles; y = 22.53-56.81x, r2 = .333), EMDI (open squares), and EMDR (open triangles) groups were similar in agreement with the consistent level of cardioprotection observed in these dogs. LV area indicates left ventricle area; AAR, anatomic area at risk.
Discussion
In the current study, we demonstrate that treatment with the NHE1 inhibitor EMD 87580 either before acute regional ischemia or before reperfusion did not protect against development of tissue necrosis. As expected, first- and second-window IC significantly reduced infarct size (cf. Figure 6) caused by 90-minute acute coronary occlusion followed by reperfusion. 8 However, when combined with either first- or second-window IC, EMD 87580 given before ischemia, or reperfusion, did not provide greater protection against ischemia–reperfusion injury; this is contrary to previously reported findings in a similar experimental model using a different NHE1 blocker (BIIB 513). 15 On the other hand, treatment with EMD 87580 did not lessen the degree of cardiac protection afforded by either first- or second-window IC.

Myocardial infarct size (% AAR) for no ischemic conditioning (noIC) compared to first (FWIC)- and second-window (SWIC) ischemic conditioning treatments. Results are means ± 1 SD. Results are means ± 1 SD, *P ≤ .05 vs no IC. AAR indicates anatomic area at risk.
Ischemic Conditioning
Tolerance to ischemia-induced myocardial necrosis has been documented using a host of pharmacologic and nonpharmacologic interventions; preconditioning-mediated cardioprotection is also effective against severe postischemic ventricular dysrhythmias 32,33 and endothelial dysfunction. 34 Later studies showed that protection was possible when the conditioning stimulus was performed 24 to 48 hours prior to a sustained coronary event. 8,9,35–39
Implication of NHE1 in Ischemia–Reperfusion Injury
Sodium–hydrogen exchange is the major cellular extrusion pathway for protons and is critical for regulation of intracellular pH 40 ; 8 different NHE isoforms have thus far been identified in mammalian cells. 41 Normally, NHE1 extrudes H+ in exchange for Na+ but with prolonged ischemia NHE1 activity is inhibited by intracellular acidosis. 42 A window of opportunity for cytoprotection exists during reperfusion after which acidic metabolites and carbon dioxide accumulate intracellularly; this leads to higher Na+/Ca2+ exchanger activity, increased Ca2+ uptake 43 –45 , and increased potential for cardiac arrhythmias, contractile stunning, and irreversible cellular injury. 46,47
Blockade of NHE1 limits development of tissue necrosis and its consequences in diverse animal models 48 –50 ; dosage and timing of administration of these drugs appear to be important factors. One particularly important protective mechanism involves attenuation of oxidative stress. 51,52 Reactive oxygen species (ROS) stimulate upstream kinases that activate NHE1 and increase intracellular Ca2+ levels; 53 both ROS and increased Ca2+ regulate opening of the mitochondrial permeability transition pore, a key contributor to ischemia–reperfusion injury. 54 –57 The NHE1 blockers and IC both reduce production of ROS 58,59 ; NHE1 blockers also preserve mitochondrial integrity during ischemia. 60 Fantinelli and coworkers suggested that by EMD 87580 and IC share common actions through a ROS-dependent pathway to protect ischemic myocardium. 61
Controversy persists regarding the critical window of opportunity for administration of NHE1 blockers; although protection afforded by these drugs administered either before ischemia 62 –67 or reperfusion 64,65,68 in different ex vivo and in vivo models has been documented, successful outcomes remain inconsistent. Interestingly, protection afforded by NHE1 inhibition alone is alleged to be better than that supplied by IC alone, 15 but findings of the present study are not consistent with this affirmation. A recent study by Sasamori et al reported marked dose-dependent reduction in myocardial infarct size in dogs (90-minute ischemia and 300-minute reperfusion) with TY-51924 (selective NHE1 blocker) administered before reperfusion. 69 Maximum protection was provided when TY-51924 was given continuously before reperfusion and maintained thereafter. While the reasons for these different results are not clear, it is possible that distinct enzyme or receptor affinities of NHE1 blockers are responsible for the diverse results. Corvera and coworkers showed that EMD 87580 conferred optimal cardioprotection when the drug was administered directly to the heart and systemically in the initial stages of reperfusion 31 ; they indicated that protection was related to timing of administration as well as dosage of this drug and suggested that higher drug concentrations were needed to afford beneficial effects. Indeed, EMD 87580 is selective but less potent than other NHE1 inhibitors. 70
Ischemic conditioning could attenuate intracellular acidosis by increasing activity of the NHE1. 71,72 Indeed, organ conditioning combined with NHE1 blockade may afford greater than additive protection 15 ; again results of the present study do not confirm those findings. On the other hand, combined IC (either first or second window) and NHE1 blockade did not diminish the ceiling of protection.
Whether NHE1 blockers influence myocardial perfusion after ischemia–reperfusion injury has not been clearly established; however, better perfusion of myocardium within the infarct area would have a positive impact on cell survival, since infarct expansion is heavily influenced by no reflow. 73 Wu and coworkers documented improved vital organ blood flow after acute hemorrhage in pigs treated with the selective NHE1 blocker, BIIB513. 74 In the present study, we report a progressive loss of vascular reserve in the infarct-related artery during reperfusion that is probably due to endothelial injury; EMD 87580 did not positively affect vascular reserve or influence CVC whether administered before ischemia or before reperfusion. We were also unable to demonstrate a substantial increase in transmural myocardial perfusion for either treatment group after restoration of flow in the infarct-related artery; at the same time, no change in transmural blood flow was detected in the adjacent nonischemic myocardium. The same results were obtained for the first- and second-window IC studies; as such, we were unable to validate earlier conclusions regarding influence of either NHE1 blockers 74 or IC 75 on postischemic tissue perfusion.
Limitations
There are some limitations in our study. Experiments were done in open chest, pentobarbital anesthetized dogs; while it is possible that anesthesia could influence experimental findings, all dogs were treated similarly for the acute ischemia–reperfusion interventions. A priori consideration was not given to the prospect of anesthetic preconditioning particularly in dogs assigned to Protocol C; these experiments were performed closed chest and therefore might have benefitted from a shorter anesthetic period (compared to earlier studies using open-chest preparations 8 ). For IC, we used the classical model described by Murry and coworkers 7 with the exception that ischemic duration was extended to 90 minutes; findings with this duration of ischemia are variable in canine experimental preparations. 8,15 Other important risk factors that can affect infarct development such as duration and depth (ie, residual flow deficit in the ischemic zone) of ischemia and anatomic risk zone size were evaluated in each animal. 40,41 In the present study, 46 male and 29 female dogs were randomly assigned to the treatment groups (each comprised at least 2 females). Gender-related differences may affect cardiac sensitivity to ischemic injury, 76,77 but we did not consider sex as an independent variable. Our findings are comparable to earlier studies in dogs regarding the development of ischemic injury. 78,79
The NHE activity was not evaluated in these studies; however, selectivity of EMD 87580 for the NHE1 isoform and the existence of this isoform (Accession # NM_0012887028) in canine tissues have been documented. 25 The cDNAs for the various NHE isoforms have been described. 80,81 The question of pharmacologic dosage for EMD 87580 merits consideration; herein, drug was given at a dose that has been reported to provide a beneficial effect in dogs. 31 Plasma EMD 87580 levels >300 to 500 ng/mL (ie, above the therapeutic range; data on file at Merck KGaA, Germany) were achieved within 5 minutes after administration and were maintained thereafter; at these plasma levels, the drug should have been able to rescue ischemic myocytes from terminal necrosis. Optimal timing, dosage, plasma concentration, and mode of drug delivery also contribute markedly to potential efficacy; this was clearly shown by Sasamori et al in their study. 69 Bioavailability within the first minutes of reperfusion is critical to affect the following factors: 1—change in cellular Ca2+ homeostasis, 2—control of intracellular pH postreoxygenation of ischemic myocytes, and 3—signaling pathways that involve Ca2+ equilibrium or myofibrillar Ca2+ sensitivity. 82 Lack of presence of the inhibitor in the early minutes of ischemia or reperfusion markedly reduces the likelihood of a beneficial effect. 83
Conclusion
The current study shows that NHE1 inhibition with EMD 87580 did not reduce infarct size when administered before ischemia or reperfusion. As such, these findings do not support the hypothesis that NHE1 inhibition reduces tissue injury produced by acute coronary occlusion. On the other hand, treatment with EMD 87580 did not antagonize protection afforded by first- or second-window IC; moreover, greater than additive cardioprotection was not observed. Based on these findings, the value of NHE1 blockers for myocardial protection against ischemia–reperfusion injury remains to be established; however, the efficacy of both first- and second-window IC is confirmed.
Footnotes
Acknowledgments
EMD 87580 was a generous gift from Merck KGaA (Darmstadt, Germany); plasma EMD 87580 levels in blood samples were also analyzed at Merck KGaA. The author would like to thank Professor Chantale Simard, Faculty of Pharmacy, Laval University, for helpful suggestions in the preparation of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the Heart and Stroke Foundation of Canada.