
Abstract
Introduction:
Dabigatran etexilate is an oral direct thrombin inhibitor. Dabigatran excretion is 80% renal, so exposure increases with severity of renal failure. The US Food and Drug Administration-approved dabigatran etexilate 75 mg twice daily (BID) for patients with nonvalvular atrial fibrillation (NVAF) having severely impaired renal function (creatinine clearance: 15-30 mL/min), based on post hoc pharmacokinetic modeling. We assessed dabigatran exposure at trough and peak levels in patients with NVAF and severely impaired renal function and compared with model predictions.
Methods:
Patients received dabigatran etexilate (75 mg BID) for ≥7 days before blood sampling; Cpre,ss (steady-state predose concentration; trough) was taken 10 to 16 hours postdose (prior to next dose), and C2,ss (steady-state concentration; peak) was taken 2 hours (± 30 minutes) postdose. Pharmacodynamic parameters at baseline (Ebase), trough concentrations (Epre,ss), and peak concentrations (E2,ss) were assessed by established coagulation assays.
Results:
Of the 150 patients screened, 60 were treated, of which 40% were male and 78.3% were white; median age was 84 years. Cpre,ss values (n = 51) were close to pharmacokinetic modeling predictions with a geometric mean (gMean) of 155 ng/mL, geometric coefficient of variation (gCV) of 76.9%, and range of 15.6 to 498 ng/mL. The C2,ss values (n = 59) had a gMean of 202 ng/mL, gCV of 70.6%, and range of 42.0 to 680 ng/mL. Pharmacodynamic effects on coagulation paralleled dabigatran concentrations. Eleven (18.3%) patients had ≥1 adverse event (AE); pharmacokinetic results for these patients versus those without AEs (n = 49) were Cpre, ss: gMean = 206 versus 145 ng/mL, gCV = 64.0% versus 78.3%; C2,ss: gMean = 243 versus 193 ng/mL, gCV = 68.9% versus 70.8%. All bleeding events (8 events in 5 patients) were considered minor by the investigators.
Conclusion:
Dabigatran exposure levels largely confirmed earlier pharmacokinetic predictions, supporting the use of dabigatran etexilate 75 mg BID in patients with NVAF and severely impaired renal function. Pharmacodynamic results were also in agreement with earlier studies. Trial Registration: ClinicalTrials.gov NCT01896297.
Keywords
Introduction
Dabigatran etexilate is an oral direct thrombin inhibitor that is approved for use to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation (NVAF) and for the treatment and prevention of venous thromboembolism in at-risk patients. 1 Approximately, 80% of circulating dabigatran is excreted via the kidneys; 1 therefore, exposure to dabigatran is higher with greater severity of renal dysfunction. 2 As a result, patients with severely impaired renal function, defined as having a creatinine clearance (CrCl) rate of 15 to 30 mL/min, or on dialysis, were excluded from phase III clinical studies 3 (supplemental material 4 ). 3
Based on the results from pivotal clinical trials, dabigatran etexilate (150 mg twice daily [BID]) was approved by the US Food and Drug Administration (FDA) for use in patients with CrCl ≥30 mL/min. 3 Dabigatran etexilate is also FDA-approved (all in patients with a CrCl rate >30 mL/min) for the treatment of deep venous thrombosis (DVT) and pulmonary embolism (PE) in patients who have been treated with a parenteral anticoagulant for 5 to 10 days (at 150 mg BID); at 150 mg BID to reduce the risk of recurrence of DVT and PE in patients who have been previously treated; and for the prophylaxis of DVT and PE in patients who have undergone hip replacement surgery, at a dose of 110 mg the first day and then 220 mg once daily. For patients with NVAF and severely impaired renal function, whose CrCl rate was in the range of 15 to 30 mL/min, a dose regimen of 75 mg BID was approved by the FDA based on pharmacokinetic modeling and simulation. 1,5,6 Subsequently, a study was conducted to assess the pharmacokinetics and pharmacodynamics of dabigatran etexilate (75 mg BID) in a small number of patients with severely impaired renal function, who had been receiving either aspirin or a vitamin K antagonist (for any indication) and found reasonable agreement between dabigatran exposure levels and modeling predictions in these patients. 7
The current study was a phase IV clinical trial, designed to characterize the pharmacokinetics of dabigatran etexilate 75 mg BID in patients with NVAF and severe renal disease/impairment (CrCl: 15-30 mL/min) who require anticoagulation therapy. This study enhances the pharmacokinetic insights of Kooiman et al 7 who looked at a similar patient group, by addressing a larger population of patients with severely impaired renal function and by examining pharmacokinetic and pharmacodynamic outcomes according to additional factors. These include an assessment of the potential impact that coadministration with either amiodarone or proton-pump inhibitors (PPIs) may have on the pharmacokinetic and pharmacodynamic parameters of dabigatran in this patient population, since these common medications have been reported to influence the pharmacology of dabigatran etexilate. 4 However, this initial examination did not include any formal hypothesis testing. The current study also compares the plasma concentration values observed in this patient population with predictions based on the pharmacokinetic model reported by Liesenfeld et al. 8
Materials and Methods
Study Design
This study is a phase IV, prospective, single-arm, open-label pharmacokinetic trial designed to assess dabigatran exposure in patients with NVAF and severely impaired renal function (defined as creatinine clearance of 15-30 mL/min calculated by the Cockcroft-Gault formula) 9 who were treated with dabigatran etexilate (PRADAXA, Boehringer Ingelheim Pharmaceuticals, Inc, Ridgefield, Connecticut) 75 mg BID for at least 7 days. This study is registered at Clinicaltrials.gov under the identifier NCT01896297. 10 The trial involved 4 separate patient visits, with the corresponding time points and key events for each visit detailed in Figure 1.
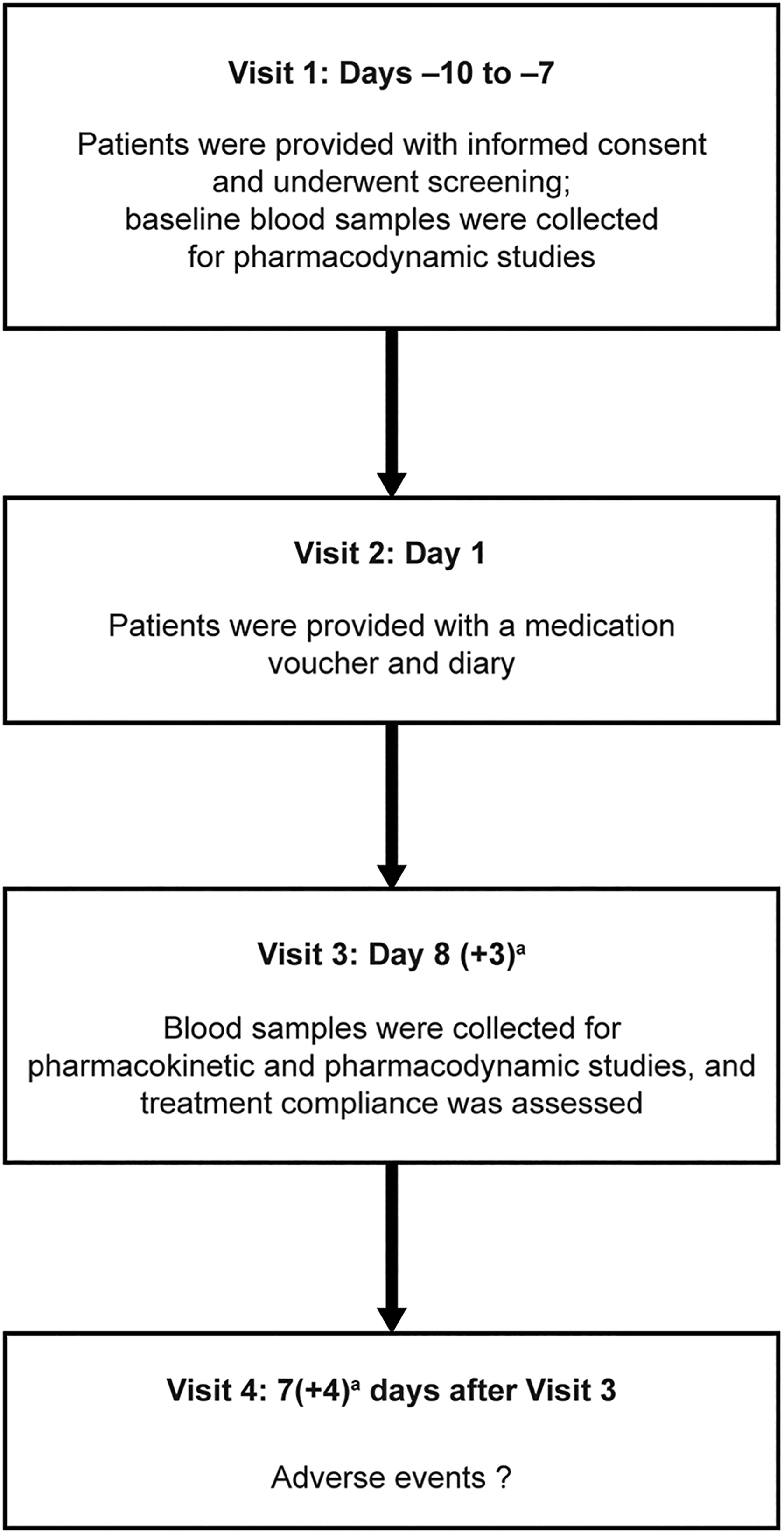
Study design.aWindow for patient visit.
The primary objective of the study was to assess dabigatran exposure at the trough, and expected peak concentrations when patients with NVAF and severely impaired renal function were dosed at 75 mg BID. The first of these measurements was the predose concentration at steady state (Cpre,ss), representing the trough concentration, taken 10 to 16 hours after the most recent dose (when the expected steady-state analyte concentration in plasma would be at the lowest concentration, immediately before the administration of the next dose). The second measurement was the concentration at steady state, peak (C2,ss), representing the expected peak concentration, taken 2 hours (± 30 minutes) after the last dose.
A secondary objective was to assess the predictability of dabigatran plasma levels based on pharmacokinetic modeling in the population of patients with NVAF and CrCl 15 to 30 mL/min by comparing the observed dabigatran trough plasma levels at steady state with those predicted by an existing pharmacokinetic model. 8 Other secondary objectives included the assessment of changes in the pharmacodynamic coagulation parameters at the baseline (visit 1), the predose, the 2 hours postdose time points at visit 3, and their relationship to the observed pharmacokinetic parameters. Pharmacokinetic results from patients who were receiving concomitant medications (ie, amiodarone and PPIs such as pantoprazole, which have been reported to influence the bioavailability of dabigatran but not to an extent that would require dose adjustment 1 ) were compared in an exploratory manner with those from patients who were not receiving the same drugs. The concomitant medication groups were not mutually exclusive.
Patients
Patients with NVAF and severely impaired renal function (enrolled set, those who signed informed consent) were enrolled with the expectation that they would complete the 4 visits with evaluable data through to the end of the treatment phase of the trial (treated set, enrolled patients who were eligible to enter the trial and received at least 1 dose of the trial drug). The inclusion criteria for this trial included both male and female patients who were at least 18 years old and who had an indication for anticoagulation therapy and severe impairment of their renal function (CrCl: 15-30 mL/min). The exclusion criteria for this trial included patients with contraindications to dabigatran etexilate (ie, serious hypersensitivity reactions), patients with active pathological bleeding, those with a mechanical prosthetic heart valve, or patients with CrCl <15 mL/min or >30 mL/min at the screening visit.
To ensure that plasma samples would be obtained from patients whose dabigatran concentrations were at steady state, treated patients must have been on the dabigatran etexilate 75 mg BID regimen for at least 7 days prior to visit 3.
Pharmacokinetic and Pharmacodynamic Assays
Dabigatran concentrations for the pharmacokinetic studies were measured in the patient plasma samples using a validated high-performance liquid chromatography–tandem mass spectrometry assay at NUVISAN GmbH (Neu-Ulm, Germany).
The activated partial thromboplastin time (aPTT), ecarin clotting time (ECT), and diluted thrombin time (dTT) assays for pharmacodynamic analyses were performed at Menal GmbH (Emmendingen, Germany), which has Good Laboratory Practices certification.
Safety Assessments
Safety analyses were descriptive in nature and based on data from all patients who were treated with at least 1 dose of dabigatran etexilate within the treatment period plus an additional 3 days to encompass the residual effect period of dabigatran etexilate.
Compliance With Ethics Guidelines
The present trial was approved by all appropriate national regulatory authorities and ethics committees of the participating centers in accordance with the principles of the Declaration of Helsinki and in accordance with the International Council for Harmonisation (ICH) Harmonised Tripartite Guideline for Good Clinical Practice. Each individual center used their own institutional review board. All patients provided written informed consent in accordance with all local regulatory and legal requirements prior to the initiation of any trial-related procedure.
Statistical Analysis
All data analyses were exploratory in nature and were assessed using descriptive statistics; N, arithmetic mean, standard deviation (SD), minimum, median, maximum, arithmetic coefficient of variation, geometric mean (gMean), and geometric coefficient of variation (gCV) were calculated for pharmacokinetic and pharmacodynamic parameters.
The predictability of dabigatran trough plasma levels was assessed based on simulations from the pharmacokinetic model. 8 These simulations were generated with the inclusion of interpatient variability, in a manner similar to the approach of Kooiman et al. 7 The main comparison between observed and predicted exposure was based on graphical representations for the 10th, 50th, and 90th percentiles of the distribution of trough concentrations. In order to obtain stable values for the concentration percentiles, the percentiles were calculated over a large number (5000) of simulation replicates.
Results
Patients
Patient enrollment reached 150 patients, 63 of which were eligible and entered the study; 3 of these patients withdrew before treatment began, 1 because of withdrawn consent, and 2 because of adverse events (AEs; 1 because of epistaxis and 1 because of an acute cerebrovascular accident). Therefore, 60 were treated with dabigatran etexilate according to the study design. One patient, an 87-year-old male, expired due to sudden cardiac death during the post-treatment phase, prior to the termination visit (Figure 2). This event was not considered to be related to the study drug.

Patient disposition. a1 patient died in the posttreatment phase and did not complete the termination visit.
Patient demographics and characteristics were generally as expected for patients with NVAF and severely impaired renal function. The majority of patients were female (60.0%) and white (78.3%). Blacks/African Americans represented 15.0% of the study patients. Patient mean age was 83.4 (± 7.55) years, and their body mass index (BMI) was 24.9 (± 4.29) kg/m2 (values presented as mean [± SD]). The time since diagnosis of NVAF was 39.7 (± 49.3) months, and the time since diagnosis of renal impairment was 40.9 (± 38.6) months (time presented as mean [± SD]). The majority of patients (56.7%) were already receiving dabigatran etexilate at baseline.
Less than half of the patients in the study had previously experienced the following conditions: gastrointestinal bleeding or ulcerative gastrointestinal disease (15.0%), stroke (20.0%), transient ischemic attack (10.0%), diabetes mellitus (40.0%), or myocardial infarction (30.0%). The majority of patients had documented heart failure (58.3%) and/or hypertension (96.7%), while their CHADS2 score was 3.33 (± 1.19) and their CHA2DS2-VASc score was 5.22 (± 1.46) (mean [± SD]).
Dabigatran Exposure: Pharmacokinetics
The pharmacokinetic standard calibration curve for dabigatran ranged from 1.0 to 400 ng/mL and was linear over the full calibration range in plasma. No evidence of interference from endogenous compounds was noted in the assay used in this study. The noncompartmental parameters for Cpre,ss and C2,ss are listed in Table 1.
Pharmacokinetic Parameters After At Least 7 Days of Dabigatran Etexilate 75 mg BID in Patients With NVAF and Severely Impaired Renal Function.

Abbreviations: BID, CrCI, creatinine clearance, twice daily; Cpre,ss, concentration at steady-state predose, trough; C2,ss, concentration at steady state, peak; gCV, geometric coefficient of variation; gMean, geometric mean; NVAF, nonvalvular atrial fibrillation; SD, standard deviation.
The results of the pharmacokinetic analysis, both at the trough (10-16 hours postdose) and at the peak (2 hours postdose) time points, were generally as expected. Over all patients in the study, the Cpre,ss concentration was 155.0 ng/mL (76.9%), and the C2,ss concentration was 202.0 ng/mL (70.6%; gMean and gCV, respectively). As also expected, in each patient group, the C2,ss concentration was numerically greater than the corresponding value reported at the Cpre,ss time point (Table 1). The observed pharmacokinetic parameters were mostly within the range of the modeling predictions. The pharmacokinetic model demonstrated a small trend toward underprediction of median and higher concentrations, while the 10th concentration percentile was slightly overpredicted. The observed and predicted (95% prediction interval) percentiles of Cpre,ss (ng/mL) were 56.3 (observed) and 76.2 (61.3-92.3; predicted) for the 10th percentile, 161 (observed) and 143 (123-166) (predicted) for the 50th percentile, and 320 (observed) and 269 (224-332; predicted) for the 90th percentile.
The potential influence of concomitant medications on the pharmacokinetic parameters was also addressed in this study (data not shown). When the pharmacokinetic parameters were examined in patients who were co-medicated with amiodarone, no apparent influence on dabigatran concentrations was observed, while for the patients who were co-medicated with PPIs, there were apparent decreases in the reported dabigatran concentrations.
Relationship Between CrCl at Baseline and Steady-State Predose Concentration
As expected, there was a general trend toward higher dabigatran concentrations with lower CrCl values. When the population was divided into subgroups based on the furthest ends of the CrCl interval (ie, CrCl 15-20 mL/min [n = 9] and 25-30 mL/min [n = 23]), the Cpre,ss medians were 213 and 146 ng/mL, respectively.
Dabigatran Exposure: Pharmacodynamics
After a minimum of 7 days of a regimen of dabigatran etexilate 75 mg BID, results for the standard coagulation assays (aPTT, ECT, and dTT) demonstrated modest elevations in reported coagulation times from baseline (visit 1) to predose (visit 3) to 2 hours postdose (also at visit 3; data not shown). It is important to note that the “baseline samples” (Ebase) included plasma samples from both dabigatran treatment-naive and pretreated patients (the latter already on the dabigatran etexilate regimen at the beginning of the study). Therefore, in this collection of all samples, the mean baseline values include those from the dabigatran pretreated patients (patients already taking dabigatran at the start of the trial, and who may have had existing plasma dabigatran concentrations at the pretreatment baseline time point). As expected, when separated by treatment status, only the data from the treatment-naive patients displayed an elevation from baseline (Table 2).
Pharmacodynamics: Coagulation Times at Key Time Points According to Dabigatran Etexilate Treatment Status.

Abbreviations: aPTT, activated; dTT, dilute thrombin time; ECT, ecarin clotting time; s, seconds; SD, standard deviation; Ebase, at baseline; Epre,ss, trough concentrations; E2,ss, peak concentrations; TT, thromboplastin time.
an displayed is maximal n; Epre,ss and E2,ss were generally calculated with a slightly smaller n. In the case of TT, Epre,ss, and E2,ss were calculated with n = 7 and 6, respectively, for treatment-naive patients.
bValues presented are arithmetic means.
The time-effect profiles of dTT, aPTT, and ECT assays at the baseline, predose, and 2 hours postdose time points are presented in Figure 3A for the treatment-naive patients and Figure 3B for the dabigatran etexilate pretreated patients.

Arithmetic mean (± SD) time-effect profiles of dTT, aPTT, and ECT after multiple administrations of dabigatran etexilate 75 mg BID in patients with severely impaired renal function (CrCl: 15-30 mL/min) at −12 hours (baseline, visit 1), −1 hours (predose, visit 3), and 2 hours (2 hours postdose, visit 3). Data are presented according to the patient groups: (A) treatment-naive patients, (B) pretreated patients. The time point of −12 hours (baseline) refers to visit 1, and the time points of −1 and 2 hours refer to the planned times for pharmacodynamic sampling at visit 3. aPTT, activated partial thromboplastin time; BID, twice daily; dTT, diluted thrombin time; ECT, ecarin clotting time; SD, standard deviation.
The clotting assay results demonstrated good correlations between measured plasma concentrations and measured laboratory coagulation parameters (Figure 4, baseline clotting times from dabigatran etexilate pre-treated patients excluded), with linear responses reported for the ECT and dTT assays, which demonstrated regression coefficients of determination (R2) of 0.91 and 0.87, respectively. The responses for the aPTT assay were curvilinear, with limited sensitivity at higher dabigatran concentrations.
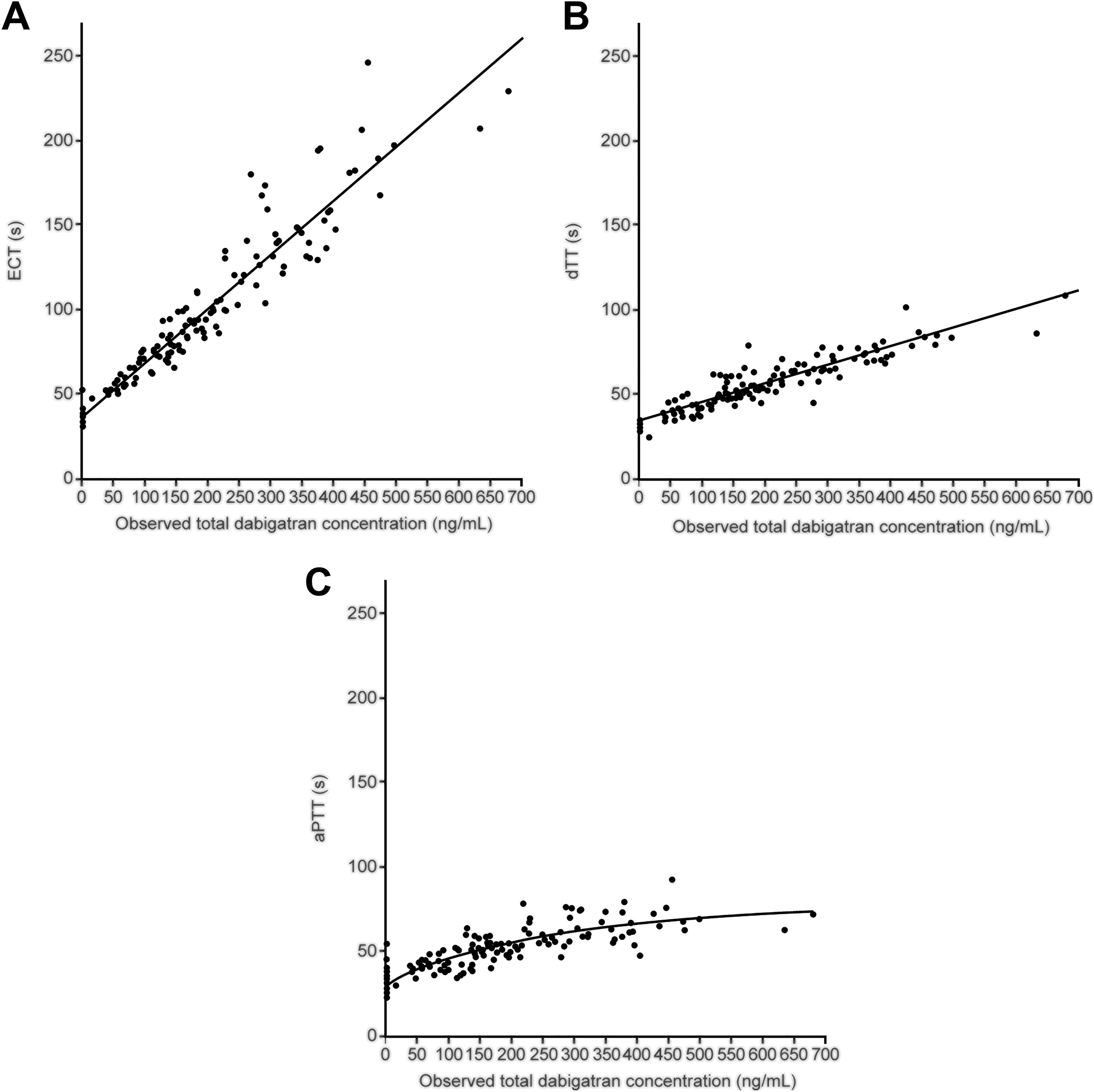
Dabigatran plasma concentrations versus laboratory coagulation parameters. Dabigatran plasma concentrations and coagulation assay results were approximately linear for ECT and dTT assays (R2 statistic = 0.91 and 0.87, respectively), and curvilinear for the aPTT assay. aPTT activated partial thrombin time; dTT, dilute thrombin time; ECT, ecarin clotting time; R2, coefficient of regression.
The potential influence of concomitant medications on coagulation parameters was also examined in these patients. Results for all coagulation parameters were in line with the pharmacokinetic outcomes.
Safety
Of the 60 patients who were treated in the trial, 11 (18.3%) patients reported at least 1 AE during the period spanning treatment through 3 days after the last dose of dabigatran etexilate. Pharmacokinetic results for these 11 patients versus those without AEs (n = 49) were Cpre,ss: gMean = 206 versus 145 ng/mL, gCV = 64.0% versus 78.3%, C2,ss: gMean = 243 versus 193 ng/mL, and gCV = 68.9% versus 70.8%. Four (6.7%) patients experienced investigator defined drug-related AEs, including hematoma and gingival bleeding, lower abdominal pain and hematuria, mouth hemorrhage, and contusion (1 patient each). During the post-treatment phase, 2 patients experienced AEs that were considered by the investigator to be study drug related (anemia and contusion, 1 patient each). All of these drug-related AEs were considered by the investigator to be minor. Of the dabigatran-naive (n = 26) and pretreated patients (n = 34), 8 (30.8%) and 3 (8.8%), respectively, experienced AEs, 1 versus 0 of which were severe, and 4 versus 0 of which were deemed drug-related by the investigator.
Two (3.3%) patients experienced serious AEs (SAEs) who required hospitalization (including a patient with an acute myocardial infarction and another with hyperkalemia), both during the treatment period (including the 3 days after the final dose of study drug). All SAEs were considered by the investigator to be unrelated to the trial drug. Another patient died during the post-treatment phase, 4 days after visit 3 due to sudden cardiac death, which was determined by the investigator to not be related to the trial drug. The most common AEs were gastrointestinal disorders and were reported by 5 (8.3%) patients. No new safety signals were noted during the course of this study.
Discussion
The data from this study help to confirm the predicted plasma concentrations of one of the earlier pharmacokinetic models used to propose dosing with dabigatran etexilate 75 mg BID in patients with NVAF and severely impaired renal function. 8 This is an important finding since there are limited pharmacokinetic data regarding the use of this dosing regimen in patients with severely impaired renal function. 7
The observed pharmacokinetic parameters were mostly within the range of the modeling predictions, and the observed ranges for the Cpre,ss and C2,ss values, while apparently large, were consistent with those previously reported. 8 Of note, the Cpre,ss values reported here are also in the same range as the Cpre,ss values measured in patients with moderate chronic kidney disease (defined as CrCl between 30 and 50 mL/min) who were treated with 150 mg BID. 11 In the patients with no major bleeds, the median (10th-90th percentiles) trough concentrations were 75.3 (30.7-175) ng/mL compared with 116 (46.7-269) ng/mL in patients with major bleeds.
The results reported in this study are also in reasonable agreement with the prediction of the model of Hariharan and Madabushi. 5 The arithmetic mean Cpre,ss observed in the present study was 187 ng/mL (n = 51), while the mean Cpre,ss predicted by Hariharan and Madabushi was ∼218 ng/mL. Similarly, the arithmetic mean C2,ss (expected maximum) observed in the present study was 242 ng/mL (n = 59), while the Cmax,ss predicted by Hariharan and Madabushi was ∼290 ng/mL.
The effects of concomitant medications on circulating dabigatran pharmacokinetic and pharmacodynamic parameters in the present patient population are consistent with those reported in the Randomized Evaluation of Long-term anticoagulant therapY (RE-LY) trial. 3 In both studies, PPI co-medication led to slight reductions in dabigatran pharmacokinetic and pharmacodynamic parameters. However, the observed reductions were not clinically meaningful, nor do they suggest a need for dose adjustment in either study. Amiodarone co-medication did not impact these parameters in either study. 3 Given the strong correlation between pharmacokinetic and pharmacodynamic parameters, the extent of the impact of co-medication on pharmacodynamic parameters was likely influenced by the sensitivity of the coagulation assay to dabigatran concentrations; thus, the changes seen in pharmacodynamics following PPI co-medication were likely a direct result of the minor changes in dabigatran concentrations. In addition, differences in the numbers of treatment-naive and pretreated patients in each of the assays would have affected the reported values at baseline.
The results described here are in line with a recent study of the pharmacokinetics and pharmacodynamics of dabigatran etexilate 75 mg in patients with severely impaired renal function, a study that involved fewer patients from a non-NVAF population, but did examine the pharmacokinetic parameters in more detail, including full temporal profiles of the measured pharmacokinetic parameters after first and last doses and an assessment of renal clearance. 7 The maximum measured concentration in plasma at steady state was 215 ng/mL (10th-90th percentiles 116-365 ng/mL).
The Cockcroft-Gault formula was used in this study to be consistent with the methodology used in the RE-LY trial. 3 This is one of several formulas available to estimate renal function, including the Modification of Diet in Renal Disease Study equation or the Chronic Kidney Disease Epidemiology Collaboration equation. 12 The Cockcroft-Gault formula remains in widespread clinical use and, therefore, can help guide understanding of the clinical status of the patients who participated in this clinical trial. Therefore, use of the Cockcroft-Gault formula to determine patient eligibility is not expected to have had a material influence on the population of patients selected for this study, and, thus, it is expected that the conclusions would remain the same regardless of formula used to estimate renal function.
When referring to oral medications, patient adherence is always a variable to be considered.
Conclusion
The main goal of this study was to help clinicians choose the right dose for this patient population. The results confirm the predictions of the earlier pharmacokinetic models used to propose dosing with dabigatran etexilate 75 mg BID in patients with NVAF and severely impaired renal function. Furthermore, the coagulation assays demonstrate pharmacodynamic responses that are consistent with the measured plasma concentrations of dabigatran. These data, along with prior studies 5 -7 focusing on patients with severely impaired renal function, support the use of the dabigatran etexilate 75 mg BID dosing regimen in this patient population.
Footnotes
Authors’ Note
The name of institution where the work reported was done: Boehringer Ingelheim Pharmaceuticals, Inc (BIPI).
Acknowledgments
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE) and were fully responsible for all content and editorial decisions and were involved at all stages of manuscript development. The authors received no direct compensation related to the development of the manuscript.
This work was supported by BIPI. Writing assistance was provided by José L. Walewski, PhD, of Envision Scientific Solutions, and editorial support and formatting assistance were provided by Simi M Chacko, PhD, and Steven Tresker, both of Cactus Communications, which was contracted and compensated by BIPI for these services.
Authors' Contribution
J. Martin contributed to conception and design, contributed to acquisition, analysis, and interpretation, critically revised the manuscript, and gave final approval. H. Esmaeili contributed to design, contributed to analysis and interpretation, critically revised manuscript, and gave final approval. R. Manuel contributed to design, contributed to analysis and interpretation, critically revised the manuscript, and gave final approval. M. Petrini contributed to interpretation, critically revised the manuscript, and gave final approval. S. Wiebe contributed to analysis and interpretation, critically revised the manuscript, and gave final approval. H. Maas contributed to design, contributed to analysis and interpretation, critically revised the manuscript, and gave final approval. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Jack Martin was the principal investigator of the “Pradaxa Study in Non-valvular Atrial Fibrillation Patients With Severely Impaired Renal Function” clinical trial, which was funded by BIPI. Habib Esmaeili is an employee of Staburo GmbH (on behalf of Boehringer Ingelheim Pharma GmbH & Co. KG). Hugo Maas and Sabrina Wiebe are employees of Boehringer Ingelheim Pharma GmbH & Co. KG. Raymond Manuel and Michaela Petrini are employees of BIPI, the manufacturer of dabigatran etexilate, and the funding source for this clinical trial.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This clinical trial was supported by Boehringer Ingelheim Pharmaceuticals, Inc (BIPI); Ridgefield, CT, USA. Dabigatran etexilate (dabigatran) was manufactured by Boehringer Ingelheim.