
Abstract
Data obtained in both preclinical models and humans have revealed that the favorable cardiac consequences of ischemic conditioning extend beyond a direct effect on the cardiomyocyte. In the current review, we summarize our as-yet limited understanding of the complex relationships between ischemic conditioning, platelet activation–aggregation, and cardioprotection.
Keywords
The Inspiration
The early 1990s were marked by a resurgence of interest in the concept of cardioprotection and a growing body of evidence that brief episodes of antecedent ischemia paradoxically “precondition” the heart and render cardiomyocytes resistant to a subsequent, sustained ischemic insult. 1 Infarct size reduction with ischemic preconditioning (PC) had been described in multiple preclinical models (reviewed in 2 –4 ), and data from clinical trials suggested that brief ischemia in the form of preinfarction angina may, similarly, be associated with a subsequent increase in ischemic tolerance of the human heart. 4 –7 Initial insights had also emerged regarding the mechanisms responsible for the reduction in infarct size achieved with PC including, most notably, the release of adenosine from ischemic–reperfused myocardium during the PC stimulus and its role as a molecular trigger for this profound cardioprotective phenomenon. 8 –10
In 1996, Andreotti and colleagues corroborated the findings of previous studies and demonstrated a robust reduction in myocardial infarct size in patients with versus without preinfarction angina. 11 Interestingly, the cohort with preinfarction angina also displayed a significant reduction in the time to achieve thrombolysis-induced reperfusion, leading Andreotti et al to propose the novel concept that the favorable cardioprotective effect of preinfarction angina “may depend on faster coronary thrombolysis, in addition to or instead of myocardial preconditioning.” 11(pp11) This seminal observation, coupled with (1) the emerging focus on adenosine as a player in PC and (2) the long-standing evidence that adenosine is a potent inhibitor of platelet activation–aggregation via stimulation of adenosine A2A receptors on the platelets’ surface, 12 –14 prompted further speculation that “adenosine, released during previous ischemia, may have influenced platelet function and favored thrombolysis.” 15(pp204)
From the Bedside to the Bench
Preconditioning Accelerates Thrombolysis
Our group was intrigued by the findings first reported by Andreotti et al 11 and replicated by others 16 and the implication that the benefits of PC may extend beyond a direct receptor-mediated effect on cardiomyocytes. Using a canine model of complete and sustained thrombotic coronary occlusion, we recapitulated and confirmed the clinical observation: The time required to achieve coronary thrombolysis with recombinant tissue-type plasminogen activator was significantly shortened in animals that received brief antecedent PC ischemia when compared to controls. 17 Moreover, and as proposed by Andreotti and colleagues, 15 adenosine appeared to play a role: The accelerated thrombolysis achieved with PC was abrogated by concurrent administration of the adenosine A2/A1 receptor antagonist CGS 15943. 17
Ischemic Conditioning Attenuates Platelet-Mediated Thrombosis: The Simple Story
Although the aforementioned results are consistent with the premise that ischemic PC (and the concomitant release of adenosine) favors thrombolysis, the data do not provide direct evidence for an effect of PC (and the concomitant release of adenosine) on platelet function. Indeed, other factors are, in all likelihood, also at play: for example, PC was associated with differences in clot architecture (in particular, greater disarray in fibrin organization) that may facilitate more rapid lysis. 18
A more direct test of the PC–adenosine–platelet function concept was conducted using the classic canine model of spontaneous, recurrent platelet-mediated coronary thrombosis. 19 –21 This model, designed to simulate the key pathophysiologic features of unstable angina, is characterized by (1) the repeated formation and dislodgment of an occlusive, platelet-rich clot (a process that is initiated by the application of a stenosis at a site of focal, experimentally induced coronary artery injury), resulting in (2) the development of cyclic variations in coronary blood flow (CFVs; Figure 1). 21 In the first series of experiments, animals were randomized to receive brief PC ischemia or a time-matched control period before the onset of LAD injury + stenosis. Per our hypothesis, LAD blood flow was better maintained (reflecting mitigation of recurrent thrombosis in the damaged and stenotic LAD segment) in the preconditioned cohort versus controls. Second, and as might be expected, brief intracoronary infusion of adenosine, administered in lieu brief antecedent ischemia, mimicked the effects of ischemic PC and improved subsequent LAD patency when compared to controls that received a matched infusion of saline. Finally, pretreatment with the adenosine A1/A2 receptor antagonist PD115,199 abrogated the favorable, inhibitory effect of PC on recurrent platelet-mediated thrombosis, thereby implicating a cause-and-effect relationship with adenosine and adenosine receptor stimulation. 21
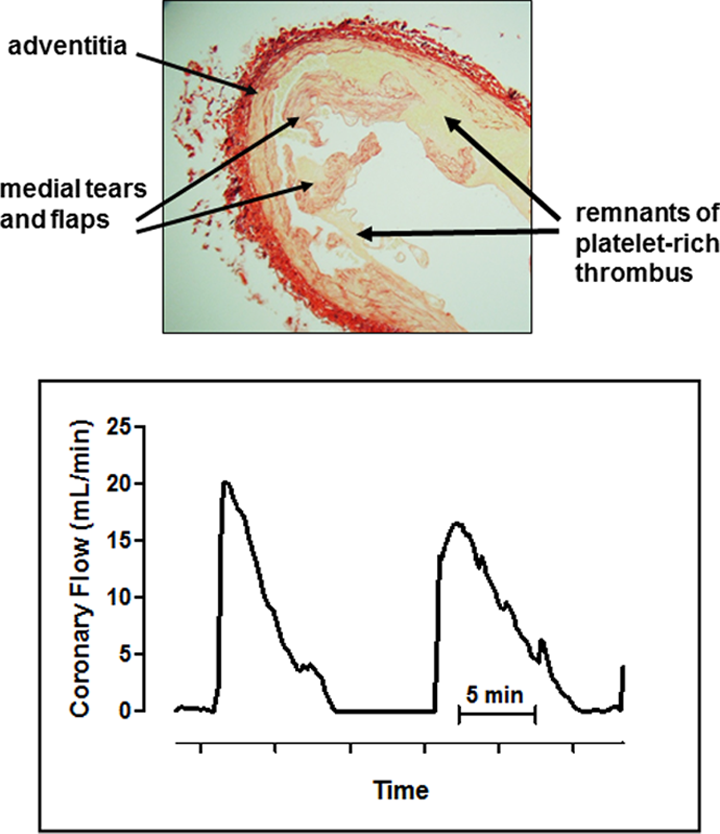
Canine model of recurrent platelet-mediated thrombosis mimicking unstable angina. Top panel: Cross-section of the damaged and stenotic segment of the left anterior descending coronary artery at the site of injury, stained with Picro-Sirius red, showing the intact tunica adventitia, tears and flaps in the tunica media, and remnants of platelet-rich thrombus in the lumen and adjacent to the vessel wall. Bottom panel: Original recording of coronary blood flow following injury + stenosis. Cyclic variations in coronary flow—the hallmark of this model—reflect the spontaneous, recurrent formation dislodgment of occlusive platelet-rich thrombi.
In all of these previous protocols, the PC stimulus was applied “locally” at the same LAD segment that served as the site of later thrombotic occlusion. 17,21 However, given the underlying premise that the favorable effects of PC on arterial patency are initiated by a humoral factor (presumably adenosine), released from ischemic-reperfused myocardium and acting on circulating blood elements (ostensibly platelets), logic would suggest that ischemic conditioning should, similarly, attenuate thrombosis at remote sites. In this regard, evidence of remote conditioning against recurrent thrombosis was obtained in the rabbit and canine models: In both species, a brief PC ischemic stimulus applied in the heart attenuated CFVs and improved patency of damaged and stenotic carotid arteries—an effect that was, again, blocked by administration of CGS 15943. 22 In addition, in all of the protocols discussed above, the local or remote conditioning stimulus was administered before arterial injury + stenosis and the onset of thrombotic occlusion. However, recent data obtained in the canine model mimicking unstable angina revealed that prophylaxis is not required, that is, remote perconditioning, with brief bilateral hind limb ischemia imposed after the onset of recurrent platelet-mediated thrombosis in the LAD coronary artery, evoked a subsequent, significant improvement in coronary patency (Figure 2). 23

Original recording of coronary blood flow following injury + stenosis, demonstrating the improvement in coronary patency seen after remote ischemic conditioning (RIC).
From Simple Story to Complex Paradigm: The “Clot Thickens”
Taken together, these studies provide a consistent body of evidence that ischemic conditioning, applied locally or remotely, improves arterial patency in models of recurrent thrombosis. These data may be interpreted to suggest that (1) coronary artery injury + stenosis, and the resultant development of CFVs, is a consequence of an upregulation in platelet activation–aggregation and (2) the better maintenance of coronary patency seen in the conditioned cohorts is explained by a favorable inhibitory effect on platelet reactivity, presumably via an adenosine A2A-mediated mechanism. Accordingly, our next priority was to interrogate these 2 as-yet unproven concepts and establish whether ischemic conditioning attenuates one or more molecular indices of platelet activation–aggregation. Using the canine model of unstable angina, data obtained by flow cytometry revealed that recurrent thrombosis in the control cohort was characterized by increases in platelet surface P-selectin expression, platelet–fibrinogen binding, formation of heterotypic (monocyte–platelet and neutrophil–platelet) aggregates, and platelet expression of von Willebrand factor, with no involvement of platelet endothelial adhesion molecule 1 or platelet glycoprotein Ib. 24 Most importantly, and consistent with our premise, the improvement in patency seen with PC was associated with significant decreases in platelet–fibrinogen binding and the formation of neutrophil–platelet aggregates (but, interestingly, not monocyte–platelet aggregates), together with a trend toward a decline in P-selectin expression, when compared to controls. 24
To address the adenosine (and, in particular, the platelet adenosine A2A receptor) component of the hypothesis, additional experiments were conducted in which animals received brief antecedent infusion of the A2A receptor agonist CGS 21680 in place of brief ischemia. As expected, the effects of CGS 21680 pretreatment on coronary patency following injury + stenosis mimicked the results obtained with ischemic PC. However—and in contrast to our hypothesis—the attenuation of recurrent thrombosis seen in CGS 21680-treated animals was not accompanied by a favorable attenuation in platelet reactivity: Formation of neutrophil–platelet aggregates, platelet P-selectin expression, and platelet–fibrinogen binding were comparable in the CGS-treated group versus controls. 25 Two additional observations corroborated this unanticipated and apparently dichotomous finding. First, in vitro experiments conducted using canine blood revealed that addition of CGS 21680 to blood aliquots attenuated platelet aggregation as assessed by whole blood impedance aggregometry but had no concomitant inhibitory effect on flow cytometric indices of platelet reactivity. In addition, in the in vivo canine model, administration of the adenosine A2A receptor antagonist ZM 241385 blocked the improvement in coronary patency, but not the favorable downregulation of platelet activation–aggregation, seen with PC ischemia. 25
The implication of these data are that—perhaps not surprisingly—the mechanisms responsible for the attenuation in recurrent thrombosis achieved with ischemic conditioning are more complex than initially thought. The improved arterial patency seen in the canine model is apparently not simply explained by release of adenosine and adenosine A2a-mediated attenuation of platelet reactivity; rather, the results suggest that multiple triggers (including adenosine and others), acting at multiple sites (possibly receptors on neutrophils as well as platelets), may be involved. In addition, evidence has emerged for the possible involvement of platelet adenosine A2B receptors in mediating and inhibiting platelet aggregation, particularly in the presence of stressors such as arterial injury. 14,26 Resolution of the molecular mechanisms by which ischemic conditioning (and adenosine) attenuate platelet activation–aggregation and recurrent thrombosis therefore remains a work in progress.
Finally, there was another interesting and unexpected layer of intricacy in this mechanistic story. In contrast to the dichotomous results obtained in the canine model, addition of the A2A agonist CGS 21680 to blood obtained from healthy adult humans attenuated both in vitro platelet aggregation and flow cytometric indices of platelet activation–aggregation. 25 That is, the mechanisms responsible for the antithrombotic effect of A2A agonist treatment appear to be species dependent—a difference that may favor possible clinical translation.
…And Back to the Bedside
While the previous study demonstrated the efficacy of adenosine A2A receptor stimulation in ex vivo human blood samples, 25 the obvious question remained: Is the favorable inhibitory effect of ischemic conditioning on platelet activation–aggregation, documented largely in the canine model, manifest in human subjects?
This issue was first addressed in healthy adult volunteers exposed to a 20-minute period of limb ischemia induced by sustained inflation of a blood pressure cuff to an occlusive pressure of 200 mm Hg—a stressor shown to be associated with the formation of heterotypic aggregates. 27,28 A remote conditioning stimulus (three 5-minute episodes of ischemia), applied to the contralateral arm at 30 minutes before the onset of sustained limb ischemia, attenuated the formation of monocyte–platelet aggregates 28 but, interestingly, had no effect on the proportion of neutrophil–platelet aggregates. 27 In addition, 3 lines of corroborative evidence have been obtained in patients with coronary artery disease. First, brief ischemia induced by low workload treadmill exercise or by a standard remote conditioning stimulus (brief intermittent arm ischemia) mitigated the increase in indices of platelet reactivity (quantified by flow cytometric assessment of monocyte–platelet aggregates and platelet GP IIb/IIIa expression) seen following subsequent and more intense exercise. 29,30 Second, remote ischemic conditioning reportedly attenuated the increase in markers of platelet aggregation accompanying invasive cardiac procedures including radiofrequency ablation of atrial fibrillation and percutaneous coronary interventions. 31,32 Finally, and arguably of greatest relevance, are the findings obtained in patients with ST-segment elevation myocardial infarction: molecular indices of platelet activation–aggregation were significantly lower in patients who experienced preinfarction angina (presumably serving as a PC stimulus) 4 –7 when compared to the no angina group. 33 All of these data provide support for the concept, first postulated on the basis of clinical observations and developed in canine models, that favorable modulation of platelet function by ischemic conditioning is manifest in patient cohorts.
Platelet Receptor Antagonists as Conditioning-Mimetic Agents?
To this point, discussion has focused on the proposed favorable consequences of local and remote conditioning on platelet reactivity—an effect which, as underscored by Andreotti and colleagues, 11,15 may make an indirect contribution, via better maintenance of coronary patency, to the overall infarct-sparing effect of this cardioprotective phenomenon. Interestingly, there is preclinical evidence that at least 1 class of potent antiplatelet drugs, currently administered as routine standard of care to patients with acute ischemic syndrome, may have direct cardioprotective effects that are purportedly not explained by inhibition of platelet-mediated thrombosis. Rather, these agents—platelet P2Y12 receptor antagonists, including clopidogrel and cangrelor—are proposed to act on cardiomyocytes and upregulate cardioprotective signaling in a manner analogous to classic ischemic conditioning. 34
The first data demonstrating infarct size reduction with P2Y12 inhibitors were obtained using the rabbit model and, subsequently, corroborated in nonhuman primates. 35,36 Moreover, in contrast to conventional ischemic conditioning, where concerns have emerged regarding loss of efficacy in the setting of clinically relevant comorbidities, 37 –39 persistent reduction in infarct size with cangrelor has been described in the Goto Kakizaki rat model of type 2 diabetes. 40 An additional and seemingly paradoxical observation has been made: Although the limitation of infarct size seen with P2Y12 antagonists is considered to be independent of their antithrombotic effects, the agents are ineffective in buffer-perfused hearts. Blood—and, specifically, platelets—is required. 35,40,41
If platelet P2Y12 receptor antagonists indeed act as conditioning-mimetic agents, this may have important implications for clinical efforts to implement ischemic conditioning as a cardioprotective intervention. Dual antiplatelet therapy, including administration of a P2Y12 inhibitor, is a cornerstone in the current treatment of patients with acute coronary syndromes. 42 The confounding effects of therapeutic strategy have been proposed to contribute to the heterogeneity in clinical outcomes obtained with ischemic conditioning (and, in particular, postconditioning). 43,44 That is, a cardioprotective phenotype may have been established by P2Y12 treatment, thereby rendering ischemic conditioning redundant and attenuating or precluding the benefit of the conditioning stimulus. 34
Next Steps
The provocative clinical observation of accelerated thrombolysis in patients with preinfarction angina, first made by Andreotti and colleagues, 11,15 provided early insight into the scope and complexity of ischemic conditioning. In addition, and in an interesting intellectual example of reverse translation, these trials served as the impetus for the design of preclinical investigations aimed at elucidating the intricate relationships between ischemic conditioning, platelets, and cardioprotection. At this point, we know that ischemic conditioning has a favorable, inhibitory effect on platelet-mediated thrombosis (which, in turn, has an indirect effect on infarct size), while administration of platelet P2Y12 receptor antagonists attenuate platelet reactivity and appear to act as direct, conditioning-mimetic agents. However, there is much we do not know. The mechanistic details of the ischemic conditioning-platelet-cardioprotection paradigm remain poorly resolved, and, perhaps most notably, the potential for the forward translation of these concepts—that is, the clinical exploitation of ischemic conditioning in the development of novel antiplatelet strategies and the targeted use of platelet P2Y12 inhibitors for clinical cardioprotection—await further study.
Footnotes
Author Contributions
Karin Przyklenk contributed to conception, drafted the manuscript, critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring itegrity and accuracy. Peter Whittaker contributed to conception, critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring itegrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received financial support for the research, authorship, and/or publication of this article: Research conducted by the authors was supported in part by NIH R01 072684.