
Abstract
A number of animal models have been designed in order to unravel the underlying mechanisms of acute ischemia-induced arrhythmias and to test compounds and interventions for antiarrhythmic therapy. This is important as acute myocardial infarction (AMI) continues to be the major cause of sudden cardiac death, and we are yet to discover safe and effective treatments of the lethal arrhythmias occurring in the acute setting. Animal models therefore continue to be relevant for our understanding and treatment of acute ischemic arrhythmias. This review discusses the applicability of the rat as a model for ventricular arrhythmias occurring during the acute phase of AMI. It provides a description of models developed, advantages and disadvantages of rats, as well as an overview of the most important interventions investigated and the relevance for human pathophysiology.
Introduction
Sudden cardiac death (SCD) caused by acute myocardial infarction (AMI) is a frequent cause of deaths in developed countries. 1 -4 Typically caused by atherosclerosis and thromboembolic events, an occlusion of a coronary artery results in complete or partial ischemia of the downstream myocardial tissue, followed by the development of an infarction. Ischemia and infarction are associated with electrophysiological disturbances both triggering and sustaining life-threatening ventricular arrhythmias, such as ventricular fibrillation (VF). Ventricular fibrillation immediately causes cardiac arrest, which is often the first symptom of AMI. 5 Ventricular fibrillation rarely spontaneously reverts to nonlethal rhythms, and among out-of-hospital cardiac arrest (OHCA) patients, only 10% to 25% survive. 6,7 Extensive clinical trials on antiarrhythmic drugs have failed to increase the survival of OHCA and thereby reduce the incidence of SCD, 8 -11 and even increased mortality with some treatments, such as flecainide and encainide in the Cardiac Arrhythmia Suppression Trial investigation, 12 has been found. Currently, the only recommended drug in the treatment of OCHA is amiodarone 13 ; however, the effect of prophylactic amiodarone on patients experiencing AMI for the first time remains unknown.
In order to enhance the treatment of ischemic arrhythmias, it is essential to understand the mechanisms underlying the development of VF. Such knowledge will provide a stronger foundation for revealing novel risk factors, as well as for selection and development of novel antiarrhythmic therapeutic strategies. An inherent problem in this undertaking is the rarity of human data on the subject. Most cases of VF induced by acute ischemia occur suddenly and unpredictably, often outside the hospital, which renders pathophysiological and electrophysiological investigation impossible. Thus, our knowledge of the time course and mechanisms behind VF development in man is very limited.
Therefore, in the past decades, a number of animal models have been developed in the attempt to mimic the process of VF caused by AMI in humans. These investigations allow control of most variables, as well as performance of invasive and sophisticated measurements unobtainable from observational and clinical studies. Consequently, investigators have gained insight into mechanisms governing VF development and can use animal models in evaluating the effectiveness of antiarrhythmic compounds.
This review will focus only on the arrhythmias occurring in the early, acute phase of AMI as these distinguish themselves from arrhythmias occurring later on (hours or days after the occlusion) with respect to electrophysiology and underlying mechanisms. 14 The focus is on in vivo rat models. For reviews on AMI models in large experimental animals, readers are referred to Hamlin 15 and Janse et al. 16 We will briefly describe the metabolic and electrophysiological mechanisms underlying VF and then the development of AMI in the rat and the rat AMI model. Following, we will review the interventions tested in these rat models and finally discuss the advantages and disadvantages of the model in relation to clinical relevance.
Mechanisms of Ventricular Tachyarrhythmias Caused by Ischemia
Occlusion of a coronary artery causes insufficient or complete loss of blood supply to the area of myocardium supplied by the artery in question, resulting in lack of oxygen and nutrients as well as accumulation of waste products. This has profound consequences for cellular metabolism and electrophysiology in this region. Decreased oxygen tension causes cardiomyocytes to shift from aerobic to anaerobic metabolism, which results in accumulation of lactate and ADP and depletion of ATP. 17 ATP shortage depresses the function of a number of ionic pumps such as the Na+/K+ ATPase and Ca2+ pumps, causing disturbances in the transmembrane gradients of several ions and substances. 18 Most importantly, Na+, Ca2+, protons, lactate, and lipid metabolites are accumulated intracellularly, whereas K+ is accumulated extracellularly. The extracellular increase in K+ occurs heterogeneously between the ischemic region, the normally perfused region, and the border zone in the middle, 19,20 causing the corresponding heterogeneities in resting membrane potential, action potential duration (APD), refractoriness, and excitability. This may result in functional obstacles and unidirectional blocks 21 which, together with nonuniform propagation of the action potential through areas of variable conduction velocities, enables initiation and propagation of reentrant circuits, which is thought to be the mechanism behind VF in the acute ischemic phase. 18
Acute Myocardial Infarction in the Rat
History of AMI in Rats
The most common technique for performing AMI in rats is by acutely tying a ligature around a coronary artery. Heimburger was the first to successfully occlude a coronary artery in rats in 1946. 22 Subsequently the model has been modified and refined, first by Johns and Olson 23 and later by Seyle 24 and Clark 25 , rendering possible the production of reliable and reproducible myocardial infarctions as well as predictable sequelae of the occlusion in regard to hemodynamics and arrhythmogenesis. The developed techniques have allowed investigators to perform large-scale testing of possible treatments, ameliorating the adverse responses of coronary artery occlusion, among these ventricular arrhythmias.
Models
The approach developed and refined by the above-mentioned researchers is performed on anesthetized rats by opening the fourth or fifth intercostal space, exposing the beating heart. At this point, respiration will cease, so the investigator can either choose to ventilate the animal with positive pressure or to perform the ligation and close the chest within a timeframe of approximately 90 seconds. 24 The pericardium is removed, and a gentle pressure on the thoracic wall exteriorizes the heart which can be held between the thumb and index finger. A suture is placed around the coronary artery in question and tightened, resulting in immediate ischemia of the downstream myocardial tissue. This is confirmed by changes in tissue coloring, wall motion, and electrocardiogram (ECG) morphology. The heart is then replaced in the thoracic cavity and the chest is closed, allowing respiration to continue.
This approach necessitates the use of anesthesia. The choice of anesthetic regimen is important, since many anesthetic agents may exert an influence over the course of arrhythmia development. Some halogenated hydrocarbons, especially halothane, has been found to possess antiarrhythmic activity in rats 26 -28 and should thus be avoided. Propofol has been reported to exhibit antiarrhythmic properties in a concentration-dependent manner, 29 presumably due to its effect on ion channels, gap junctions, and the autonomic nervous system (ANS). 30 Thiopental 31 and the combination of diazepam and ketamine 32 and ketamine and xylazine, 33 respectively, have shown antiarrhythmic effects as well, whereas urethane has been proven to both abbreviate the duration of ischemia-induced ventricular arrhythmias and cause marked depression of hemodynamic properties, 31,32 rendering this agent unsuitable for cardiovascular research in general. The most commonly used anesthetic, pentobarbital, does not mitigate arrhythmogenesis 26,31,32 and neither do anesthetics such as isoflurane, 27 sevoflurane, 29 and fentanyl. 28 These agents are therefore well suited for the study of potentially antiarrhythmic drugs.
In order to eliminate the potential confounding influence of anesthesia and acute surgical trauma, Johnston et al, 34 Lepràn et al, 35 and Himori and Matsuura 36 further developed the model, rendering possible occlusion in conscious rats. In this model, the before-mentioned surgery is still performed under anesthesia, but instead of ligating the artery, an occluder, consisting of a suture forming a snare with the ends guided through a polythene tube, is placed around it. The chest is closed with either the tubing or the ends of the suture remaining outside the body. After recovery, the snare can be tightened while the animal is conscious. This technique also allows for reperfusion by simply loosening the snare.
Comparative Anatomy of the Rat Heart and Placement of Ligation
The heart of the rat differs widely from the human heart with respect to size and heart rate. A rat heart weighs around 1.5 g and is characterized by a short cardiac cycle length with a basal heart rate of 300 to 350 beats per minute. 37 As in humans, the rat’s left ventricle is supplied by the left coronary artery (LCA) that origins from the aorta and emerges between the left pulmonary cone and the left atrial appendage. 24 The LCA branches out into a septal artery and a left anterior descending (LAD) artery. 24 In contrast to humans, there is no true circumflex artery in rats 23 and very few collateral branches.
To induce AMI in the rat, the LCA or the LAD is ligated. The place of ligation affects outcome, since the incidence of arrhythmias correlates with the size of the ischemic area. 34,38 -40 Thus, a proximal occlusion is more arrhythmogenic than a distal occlusion and will cause a higher VF incidence. Therefore, proximal ligation is often used in studies examining antiarrhythmic drug effectiveness, as a high VF incidence is necessary for detecting drug effects in small-group sizes.
During ligation, inclusion of a bundle of cardiac muscle and small veins cannot be avoided due to the small size of the coronary, but Heimburger found that tightening of the suture did not produce any significant changes unless the coronary artery was included. 22 Other methods of occlusion, such as electrocautery 41 and cryoablation, 42 have been carried out but are not common.
Time Course of Ischemia-Induced Arrhythmias
In overall terms, ischemia-induced arrhythmias can be divided into 2 phases: phase 1, which occurs during the first 30 minutes after occlusion, and phase 2, which occurs after approximately 90 minutes of occlusion, when myocardial injury is becoming irreversible. 43 In many species, phase 1 is bimodal and can be further subdivided into phase 1a and phase 1b, which differ in time course and presumably underlying arrhythmogenic mechanism. Although phase 1a arrhythmias most likely are caused by reentry, phase 1b arrhythmias probably rely on abnormal automaticity. 18 In rats, phase 1 is monomodal. It occurs during the first 30 minutes after occlusion 25,34,40 and is believed to be caused by reentry. 40 Whether phase 1 is monomodal or bimodal in humans remains to be established.
In experimental rat models, incidence of severe ventricular arrhythmias is generally high. During the first 30 minutes after occlusion of the LCA or LAD, ventricular tachycardia (VT) occurs in 73% to 100% and VF in 57% to 89% of animals. 38 Examples of these arrhythmias illustrated in Figure 1. Variations in arrhythmia incidence can be explained by differences in protocols, whether resuscitation is attempted and the place of ligation, affecting infarct size. Despite some variability between studies, the finding of 2 arrhythmogenic periods (phases 1 and 2) and a high rate of VT and VF is consistent. 40,44 -47 VT/VF incidence is much higher than reports from clinical studies, 48 -50 probably since a high control VF incidence is convenient when investigating effectiveness of antiarrhythmic drugs. Thus, rats are often selected for their low collateral flow, which in combination with a proximal ligation ensures large ischemic zones and high VF risks. Though incidence of VF is high in rat models, mortality is not, which is due to the fact that rats often spontaneously revert from VF back to sinus rhythm. 51
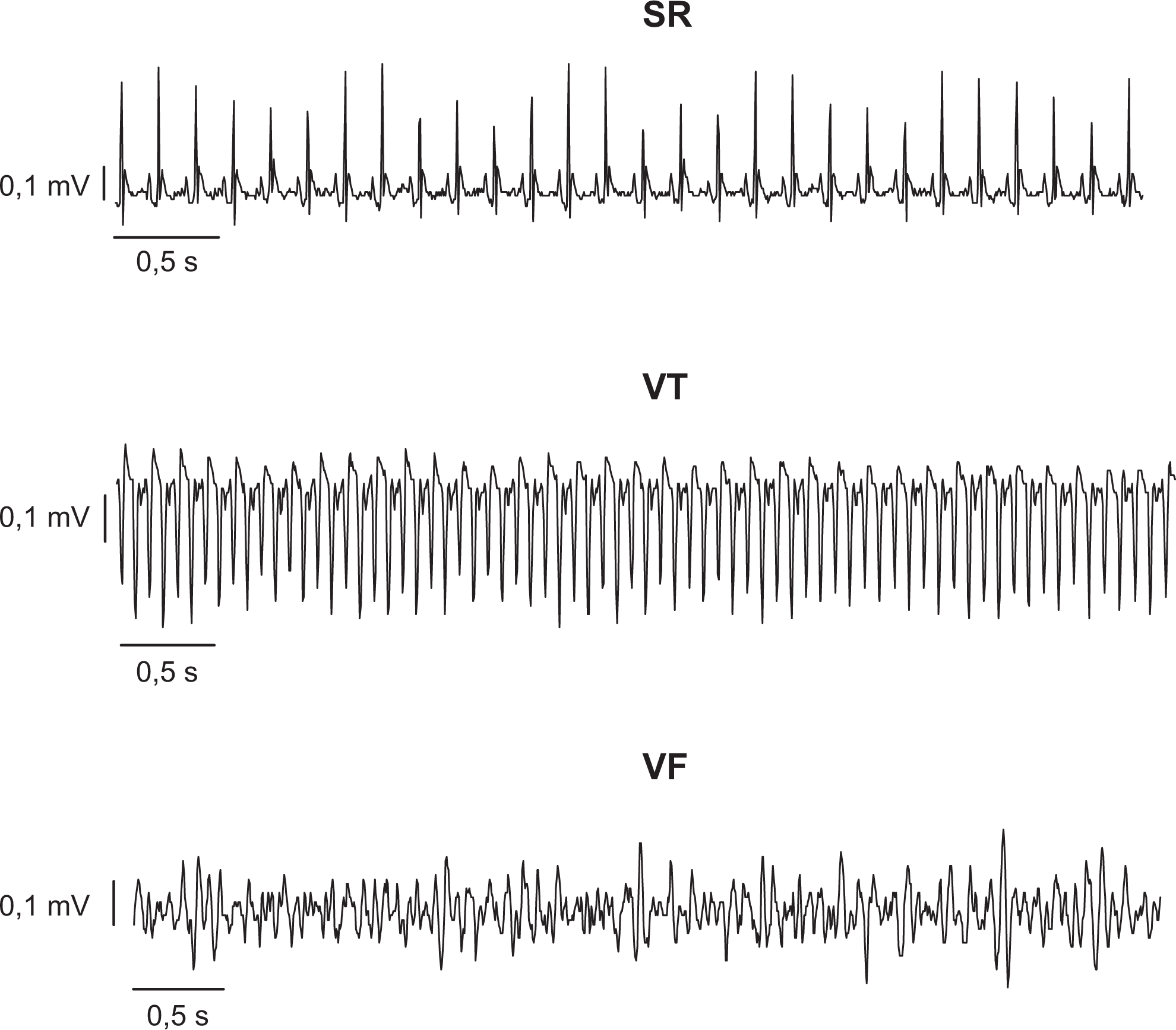
Electrocardiogram (ECG) traces of sinus rhythm (SR), ventricular tachycardia (VT), and ventricular fibrillation (VF) from rat (Hundahl, unpublished data).
Changes in ECG and Hemodynamics
Upon ligation, characteristic changes in rat ECG morphology take place. The R wave amplitude is increased immediately after ligation and soon thereafter ST segment elevation develops, whereas Q waves appear later. 34 ST elevation is a hallmark of transmural ischemia in humans as well, and cumulative ST deviation has been shown to correlate with VF risk in the clinic. 5,52 In both anesthetized and conscious rats, coronary occlusion has no profound effect on heart rate, whereas blood pressure drops within 2 minutes 25,34 and remains depressed for 24 hours in conscious rats. 34
Alternative Models
Some investigators have sought to increase the severity of ischemia-induced ventricular arrhythmias by modifying the classical rat model, reasoning that a higher incidence of easily diagnosable VF with increased mortality will improve the assessment of the antifibrillatory effect of investigated drugs while using fewer animals. Administration of isoprenaline prior to coronary ligation significantly increased VF incidence and mortality as well as the levels of significance between control and antiarrhythmic drug-treated groups. 53 In another modified model, isolation stress by single housing or chronic exposure to cold likewise increased incidence of and mortality from VF, 54 possibly due to an increased release of catecholamines and corticosteroids.
Evaluation of Arrhythmias and Changes in Electrophysiological Properties
The ventricular arrhythmias caused by myocardial ischemia can be recorded and measured in a number of ways. Typically, a lead II ECG is recorded using standard limb leads in anesthetized rats, as first described by Normann et al in 1961, 55 or telemetry devices in conscious rats 40 and used for arrhythmia analysis in accordance with the Lambeth Conventions. 56 In some studies, arrhythmias are quantified by arrhythmia scores, allowing the investigator to statistically evaluate the arrhythmic events as a collective score. Arrhythmia scoring has been reviewed and validated by Curtis and Walker. 57 More sophisticated electrophysiological measurements can also be applied to rat models. Examples are programmed electrical stimulation (PES), 58 monophasic action potentials, 59 and effective refractory period, 46 providing further insight into the underlying mechanisms of the arrhythmias present.
Interventions Applied to Rat Models of VF Caused by AMI
A great number of surgical and pharmacological interventions have been applied to the AMI rat model in order to evaluate their effect on arrhythmogenesis in the acute phase. In this study, we have chosen to review some of the most frequently tested interventions as well as some novel, promising pharmacological treatments.
Ischemic Preconditioning
Exposure of the myocardium to brief ischemic episodes has long been known to protect the heart from tissue necrosis during subsequent ischemic insults, 60 as well as the ventricular arrhythmias occurring in the setting of ischemia and reperfusion in the rat 44,61 -63 and a number of other species, including humans. 64 -66 This powerful endogenous protective mechanism is referred to as ischemic preconditioning (IPC).
IPC causes myocardial release of paracrine substances, which trigger intracellular pathways converging on end effectors that mediate the effect of IPC. Identifying the links in the IPC cascade can provide new potential targets, eventually leading to the development of pharmacological compounds mimicking the protective effect of IPC to be used in the clinic. Such investigations performed in a number of different species have led to the discovery of an extensive triggering system that includes the release of bradykinin, adenosine, oxygen radicals, opioids, and prostaglandins from the ischemic myocardium. 67 In rat, the protective effect of IPC has been shown to be impaired by administration of N-2-mercaptopropionylglycine, a reactive oxygen species (ROS) scavenger 68 and naloxone, a nonselective opioid antagonist 69 in vivo and by HOE 140, a bradykinin B(2) receptor blocker, 70 and opioid receptor antagonists norbinaltorphimine and naltrindole 71 in vitro. The role of adenosine in rats is uncertain, 72 and contradicting results have been found with regard to prostaglandins. 62,73 At least some of these triggers seem to mediate human IPC as well. For example, bradykinin is degraded by the angiotensin-converting enzyme (ACE) and administration of ACE inhibitors; increasing bradykinin levels have decreased both infarction size and arrhythmias in humans. 74
Receptor activation by triggers initiates intracellular pathways, presumably involving protein kinase C in the rat. 71,75 ATP-sensitive K+ channels (KATP) channels, especially the mitochondrial ones, have been suggested as the end effector in IPC in many species, including rats, 76 potentially explaining the antiarrhythmic effect of KATP channel openers. 45,77 -79 Some investigators found no inhibitory effect on IPC by blocking mitochondrial KATP channels, but rather by blocking the sarcolemmal channels. 70 This controversy may be due to incorrect assumptions regarding the selectivity of KATP channel openers and blockers. 80 Targeting KATP channels in antiarrhythmic therapy is discussed below.
In rats, IPC reduces dispersion in QT-interval, which is a marker for repolarization heterogeneity, and this is associated with a lower incidence of ischemic arrhythmias. 47 Increasing evidence indicates that IPC decreases cellular electrical uncoupling by preserving the function of gap junctions 81,82 and that this might be related to the antiarrhythmic effect of IPC since slow and heterogeneous uncoupling enhances the risk of reentry. 18
Ischemic Postconditioning
The phenomenon of ischemic postconditioning refers to the protective effect of brief periods of reocclusion during early reperfusion. Compared to preconditioning, which occurs prior to the permanent occlusion, postconditioning can be applied clinically in the setting of coronary reperfusion. In a number of experimental 83,84 as well as clinical studies, 85 -87 ischemic postconditioning reduced infarct size, presumably via many of the same pathways as described for IPC. 84 Though a few negative results exist, 88 postconditioning generally seems to be cardioprotective.
In rats, ischemic postconditioning have furthermore exhibited antiarrhythmic properties. In an in vivo ischemia/reperfusion model, ischemic postconditioning in the form of reperfusion/reocclusion sequences preceding final reperfusion reduced both incidence and duration of ventricular tachyarrhythmias 89,90 but failed to reduce infarct size, 90 making the authors question whether ischemic postconditioning is as straightforward as IPC in rats. Another group compared the effect of preconditioning versus postconditioning in rats under identical experimental conditions and found that preconditioning decreased infarct size whereas postconditioning did not. 91 However, both preconditioning and postconditioning protected against arrhythmias. The underlying mechanisms for the antiarrhythmic effect of postconditioning remain elusive. Blocking some pathways known to mediate IPC (p13-kinase pathway, mitochondrial permeability transition pore, mitochondrial KATP channels, and adenosine receptors) did not affect protection against arrhythmias. 92
Modulation of the ANS
AMI is associated with autonomic imbalance, 93 influencing the course of arrhythmogenesis. This process has been widely investigated in rats. Investigations of the effect of the ANS on ischemia-induced arrhythmias can be divided into 2 categories: surgical and pharmacological interventions. Surgically, nerve stimulation, ablation, and deafferentation have been attempted. Sympathetic nerve stimulation increased the incidence of ischemia-induced ventricular tachyarrhythmias and promoted dephosphorylation and degradation of connexin 43 (Cx43), 94 whereas vagal nerve stimulation exerted an antiarrhythmic effect, 95 which has been shown to be associated with prevention of dephosphorylation of Cx43. 96 Thus, it seems that autonomic tone can affect intercellular electrical coupling and thereby arrhythmogenesis. Lujan and colleagues impaired the sympathetic input to the heart by performing ablation of cardiac sympathetic neurons 97 and cardiac spinal sympathetic deafferentation 98 and found both interventions to be antiarrhythmic by decreasing sympathetic input to the heart. This is in line with clinical results, which have shown sympathectomy to be antiarrhythmic in patients with electrical storm 99 or incessant VT. 100 Curtis and colleagues performed various degrees of CNS ablations (pithing, spinalization, and decerebration) in combination with acute or chronic surgery in rats and found that all procedures involving acute surgery protected against ischemia-induced arrhythmias. 101 However, the antiarrhythmic effects did not correlate with CNS ablations, suggesting that acute surgery itself is antiarrhythmic, possibly due to changes in leucocytes, platelets, and serum K+. Additionally, heart transplantation in rats, which results in complete autonomic denervation, did not cause changes in incidence or severity of arrhythmias induced by coronary occlusion, 102 and adrenalectomy to prevent increases in plasma catecholamines did not protect against arrhythmias, 103 leading these research groups to question whether sympathetic stimulation is necessary for arrhythmogenesis in rats.
Pharmacologically, the effect of blocking or activating the different adrenergic receptors in rats is ambiguous. Although β-adrenergic stimulation by isoprenaline is proarrhythmic, 53 blocking the β-adrenergic receptors have yielded both positive 35,104 -106 and negative results. 26,107 -109 Some investigators believe that the antiarrhythmic effect of β-blocking agents sometimes observed in rats is due to blockage of cardiac-β adrenergic receptors, 105 and others have suggested that the antiarrhythmic properties are due to peripheral effects, for example, increasing levels of serum K+ 110 or neurokinin activity. 104 In humans, administration of β-blockers reduced SCD and occurrence of ventricular arrhythmias following AMI 111 -113 ; however, there are no data on the effect of prophylactic β-blockade on acute ischemic arrhythmias. In rats, administration of adrenaline, noradrenaline, and phenylephrine has been shown to protect against arrhythmia development, 53 possibly by activating the vasovagal reflex and thereby increasing vagal tone. Later studies likewise found an antiarrhythmic effect of noradrenaline, which was proposed to be due to activation of α1 receptors and mitochondrial KATP channels 44 as well as nitric oxide and ROS release. 114 Stimulation of α2 receptors has likewise been found to be antiarrhythmic. 109 Finally, increasing vagal activity by administration of the acetylcholinesterase inhibitor pyridostigmine decreased arrhythmia incidence and preserved Cx43 in AMI rats. 115
In summary, results in rats correlate well with the clinical situation as autonomic imbalance promotes arrhythmogenesis. From both clinical and experimental results, it generally seems that increasing vagal tone is protective, whereas increasing sympathetic activity is proarrhythmic. Though probably not essential for arrhythmogenesis, sympathetic overactivity seems to at least modulate the heart in a way that renders the heart more prone to arrhythmias.
KATP Channels
In the past 2 decades, a great deal of research has been conducted on the antiarrhythmic effect of KATP channel modulation in the setting of myocardial ischemia. Since these channels are activated by a decreased ATP/ADP ratio, they may represent an ischemia-selective target in the heart. It is well established that opening of KATP channels during myocardial ischemia contributes to K+ efflux, 116 causing shortening of the APD 117,118 as well as heterogeneity in repolarization 119,120 which, according to classical theory, should increase the risk of reentrant arrhythmias occurring. Thus, KATP channel blockers such as the sulfonylureas glibenclamide 77 and tolbutamide, 121 as well as the cardioselective compounds HMR 1883 122,123 and HMR 1098, 78 have been shown to possess antiarrhythmic activity in rats in vivo. Surprisingly, however, KATP channel openers in many cases have been reported to exhibit antiarrhythmic properties as well. 45,77 -79 Although blocking the KATP channels mitigates reentrant arrhythmias, opening of the channels can hyperpolarize the diastolic membrane potential, reducing intracellular Ca2+ overload, 124 thus suppressing triggered activity 125,126 as well as the degree of ischemic injury. 127 It seems that mitochondrial KATP channels also play an essential role in the antiarrhythmic and cardioprotective effect of KATP channel openers by modulating ROS production and mitochondrial matrix swelling, moderating mitochondrial Ca2+ accumulation and improving mitochondrial energy production as well as being a part of the signaling cascade of IPC. 80,128 Effects on KATP channels in other cardiovascular tissues probably also contribute to the overall picture. The KATP channel openers, pinacidil, cromakalim, and diazoxide, are known to induce vasodilation 129 which may be beneficial by decreasing cardiac afterload, causing coronary vasodilation as well as inducing baroreflex-mediated sinus tachycardia, potentially overdriving ventricular tachyarrhythmias.
Considering all data from in vivo rats, it seems that opening of KATP channels during ischemia is generally detrimental by causing reentrant arrhythmias. A number of researches have suggested that the best solution is inhibition of the sarcolemmal KATP channels and activation of the mitochondrial KATP channels, 78,130 but this requires sufficient selectivity of compounds. Several such drugs have been developed (eg, HMR 1883, HMR 1098, HMR 1402, diazoxide, and BMS-191095), but their selectivity has been questioned in a recent review by Foster and Coetzee. 80
In summary, KATP channels constitute an interesting target for antiarrhythmic therapy in the future. Sulphonylureas, which are KATP channels blockers, have long been used in the treatment of diabetes; however, so far no human data exist on the possible antiarrhythmic effect of sulphonylureas in the setting of AMI.
Small Conductance Ca2+-Activated K+ Channels
Small conductance Ca2+-activated K+ (SK) channels are expressed in a number of tissues including the nervous system, smooth muscle, epithelia, blood cells, 131 and heart. 132 In the heart, SK channel expression is more abundant in the atria than in the ventricles 132 and seems to constitute a promising target for the treatment of atrial-specific arrhythmias such as atrial fibrillation. 133,134 Under normal physiological conditions, ventricular SK channels are believed not to play a major role. Thus, application of the selective SK channel blocker apamin to healthy ventricular preparations from rats, dogs, and humans was without effect. 135 However, an increasing amount of evidence have revealed that ventricular SK channels may play an important functional role in pathophysiological conditions such as heart failure 136 and AMI and contribute to the occurrence of ventricular arrhythmias in the setting of AMI. Patch clamp studies carried out in a rabbit model of chronic MI 137 revealed that the SK current (I SK) was increased in the peri-infarcted area as compared to in a distant area and control. In the same model, optical mapping during Langendorff perfusion showed that the APD was shortened in the peri-infarcted zone compared to normal ventricles and that this could be reversed by apamin. These data indicate that increased I SK contributes significantly to ventricular repolarization in a heterogeneous manner after MI. This leads to the speculation that ventricular SK channels might also come into play in the acute phase of MI, which is characterized by, among other things, intracellular accumulation of Ca2+. 138 So far, only 1 study has investigated the effect of SK blockers on ventricular arrhythmias in acute MI in vivo. 46 Blocking the SK channels reduced the incidence and inducibility of VT and VF as well as increased VF threshold, ventricular effective refractory period (vERP), and monophasic action potential duration at 90% repolarization (MAPD90). Other investigators have found the same effect of blocking ventricular SK channels on vERP and APD ex vivo. 139 Therefore, it seems plausible that SK channels play an important role in acute MI, where Ca2+ overload and increased cytoplasmic Ca2+ release induce activation of SK channels, resulting in nonuniform increased repolarization that might serve to uphold reentry rotors and ventricular tachyarrhythmias.
In accordance with experimental results, I SK was found to be upregulated in studies examining ventricular tissue from heart failure human hearts and contribute to repolarization. 140,141 Whether the antiarrhythmic effect of SK channel inhibitors on ischemia-induced ventricular arrhythmias can be translated to humans remains to be investigated.
Connexin 43
Cx43 is the key protein making up the gap junctions of the heart, which are essential for fast, coordinated impulse conduction. Ischemia is known to cause dephosphorylation of Cx43 142 and uncoupling of gap junctions, 143 thus regionally decreasing intercellular electrical coupling, causing heterogeneities in conduction velocity, and facilitating reentrant arrhythmias. Several different investigators studying the antiarrhythmic effect of various compounds in AMI rats have discovered the antiarrhythmic effect to be associated with preservation of Cx43 and prevention of dephosphorylation. These compounds include taxol, 144 ghrelin, 145 nitrite, 146 pyridostigmine, 115 κ-opioid receptor agonists, 147 as well as nonpharmacological interventions such as IPC 81,82 and vagal nerve stimulation. 96 The antiarrhythmic effect of propofol in the AMI rat model has also been suggested to result from prevention of Cx43 dephosphorylation. 29 Thus, a number of antiarrhythmic interventions seem to converge on pathways preserving Cx43 function. Compounds directly targeting Cx43 have been developed and in rats shown to attenuate Cx43 dephosphorylation, 148 reduce infarct size, 149 prevent atrial conduction slowing during metabolic stress, 150 and protect against pharmacologically induced VF. 151 However, their effect on ischemia-induced ventricular arrhythmias has not yet been investigated in rats and could thus be an interesting future target.
Class I, III, and IV Antiarrhythmics
The classical antiarrhythmic agents classified according to Vaughan Williams have generally exhibited antiarrhythmic properties in AMI rats. However, with the exception of β-blockers and the class III drug amiodarone, most of these compounds are no longer applied in the clinic due to lack of action or adverse effects. This includes the class I drugs encainide, flecainide, 12 moricizine, 8 and lidocaine 11 as well as several class III agents 9,152 and class IV agents. 10 Therefore, results obtained in rats will only briefly be discussed due to the lack of clinical relevance.
Class I agents, which block the fast inward Na+ current, are universally antiarrhythmic in AMI rat models 25,34,51,53,153 -155 with the exception of moricizine. 156 This stands in contrast to clinical trials in which class I agents exhibit no effect on mortality or VF in patients with AMI. 8,11,12 Only a modest amount of research has been conducted on class III compounds, blocking outward K+ currents, perhaps due to the fact that rats lack the delayed rectifier K+ currents. Melperone, 157 bethanidine, meobentine, 158 sotalol, 159 and tedisamil 160 all protected against arrhythmias; however, except for tedisamil, none of these drugs are selective class III agents but also possess ancillary properties which could contribute to their antiarrhythmic effect. The antiarrhythmic effect of tedisamil can instead be ascribed to blockade of I to, a K+ current appearing early in both the rat and human ventricular AP. 161 Amiodarone, today’s drug of choice for a number of different arrhythmias, is classified as a class III drug but possesses actions similar to class Ia, II, and IV agents as well, which might account for the fact that amiodarone was found effective against ischemia-induced arrhythmias when given chronically 59,162,163 and acutely 58,164 to AMI rats. Finally, most investigators have found antiarrhythmic effects of class IV agents, blocking the inward Ca2+ current. 34,107,165 -168 However, a number of studies found no antiarrhythmic effect of class IV drugs in rats, 157,169,170 and administration of high-dose class IV drugs is associated with increased mortality due to atrioventricular conduction defects or persistent VF. 34,107,171 This is in correlation with clinical trials in which a trend toward more deaths and reinfarctions was observed for patients with MI treated with nifedipine, nicardipine, or verapamil. 10
Late Na+ Current
In cardiomyocytes, the fast upstroke of the AP is governed by a fast Na+ current mediated by Nav1.5 channels. These channels undergo rapid activation followed by rapid inactivation. Upon repolarization, the channels deactivate and can once again be activated and contribute to the upstroke of the following AP. During the plateau phase of the AP, a small fraction of the channels reactivate, resulting in a I Na, late. This late current is much smaller than the fast equivalent but has been reported to be augmented by myocardial ischemia. 172 The I Na, late contributes to Na+ loading during the cardiac cycle which may trigger arrhythmias by inducing afterdepolarizations, both early (EADs) and delayed (DADs) 172 ; increased I Na, late reduces repolarization, promoting reactivation of Ca2+ channels, and thus EADs and increased Na+ load activate the Na+/Ca2+ exchanger (NCX) to extrude Na+, causing Ca2+ influx and thus promoting DADs. Hence, increased Na+ load can cause Ca2+ overload, promoting myocardial contracture and cardiac arrhythmias by inducing EADs and DADs as well as SK channels activation (see previous section).
I Na, late is blocked by the antianginal drug, ranolazine. In rats, ranolazine reduced both ischemia- 173 and reperfusion-induced arrhythmias 173,174 and showed antiarrhythmic properties equal to lidocaine and sotalol. 175 In humans, ranolazine is widely used in the treatment of angina. Whether the drug can also find use in the treatment of ischemic arrhythmias remains to be established. In patients with non-ST elevation MI, ranolazine reduced incidences of nonsustained VT, supraventricular arrhythmias, and bradyarrhythmias, 176 suggesting an antiarrhythmic role in humans. The drug neither decreased nor increased mortality, 177 in contrast to several clinical trials on class 11,178 I, class 9,152 III, and class 10 IV antiarrhythmic drugs, which were shown to increase arrhythmias, deaths, or both. Thus, I Na, late remains an interesting target for human antiarrhythmic therapy, and further studies are warranted on its effect.
Appropriateness of Rat AMI Models
Clinical Relevance
Rat models reflect human pathophysiology in a number of parameters. Most prominently, a number of interventions have the same effect in rats and humans with AMI. For instance, increasing serum K+ 101,179 -183 and IPC 44,61 -64,66,184 is antiarrhythmic in both species. Furthermore, phase 1 arrhythmias occur spontaneously in response to occlusion in rats, mimicking the clinical situation in contrast to models in which, for example, PES is necessary to produce ischemic VF. 185 In both man and rat, phase 1 arrhythmias occur within the first 30 minutes of occlusion. 25,34,40 Finally, in vivo models closely represent clinical AMI than in vitro models; the same goes for conscious animals versus anesthetized ones.
In other aspects, the rat differs from human physiology and pathophysiology, particularly with respect to electrophysiology. In humans (and large experimental animals), the cardiac AP can be divided into 5 phases (termed 0-4) that are governed by depolarizing, inward (Na+ and Ca2+), and repolarizing, outward (K+) ionic currents (Figure 2). In rat, the voltage-gated Na+ and Ca2+ currents largely reflects the human currents. However, an early large outward K+ current results in an approximately 10-fold faster speed of repolarizing as compared to the human AP. 186 This early K+ current affects the morphology of the rat ventricular AP that distinguishes itself by the absence of a defined phase 2. 187 The swiftness of repolarization in rats, especially at high heart rates, relies almost exclusively on the transient outward K+ current (I to), 186 which later is complemented by a slower activating K+ current. 188 In humans, the rapidly activating and inactivating I to is responsible for the transient repolarization of phase 1, whereas the full repolarization of phase 3 relies primarily on delayed rectifier currents, most importantly I Ks, I Kr, and the inward rectifier current I K1. The delayed rectifier currents do not play a role in rat ventricular repolarization, as blocking these currents neither affects APD 189 nor ischemic arrhythmias, 190 in contrast to guinea pigs, rabbits, and larger animals, which are known to express these delayed rectifier K+ currents. Thus, the rat model is not suited for evaluation of drugs targeting the delayed rectifier currents.
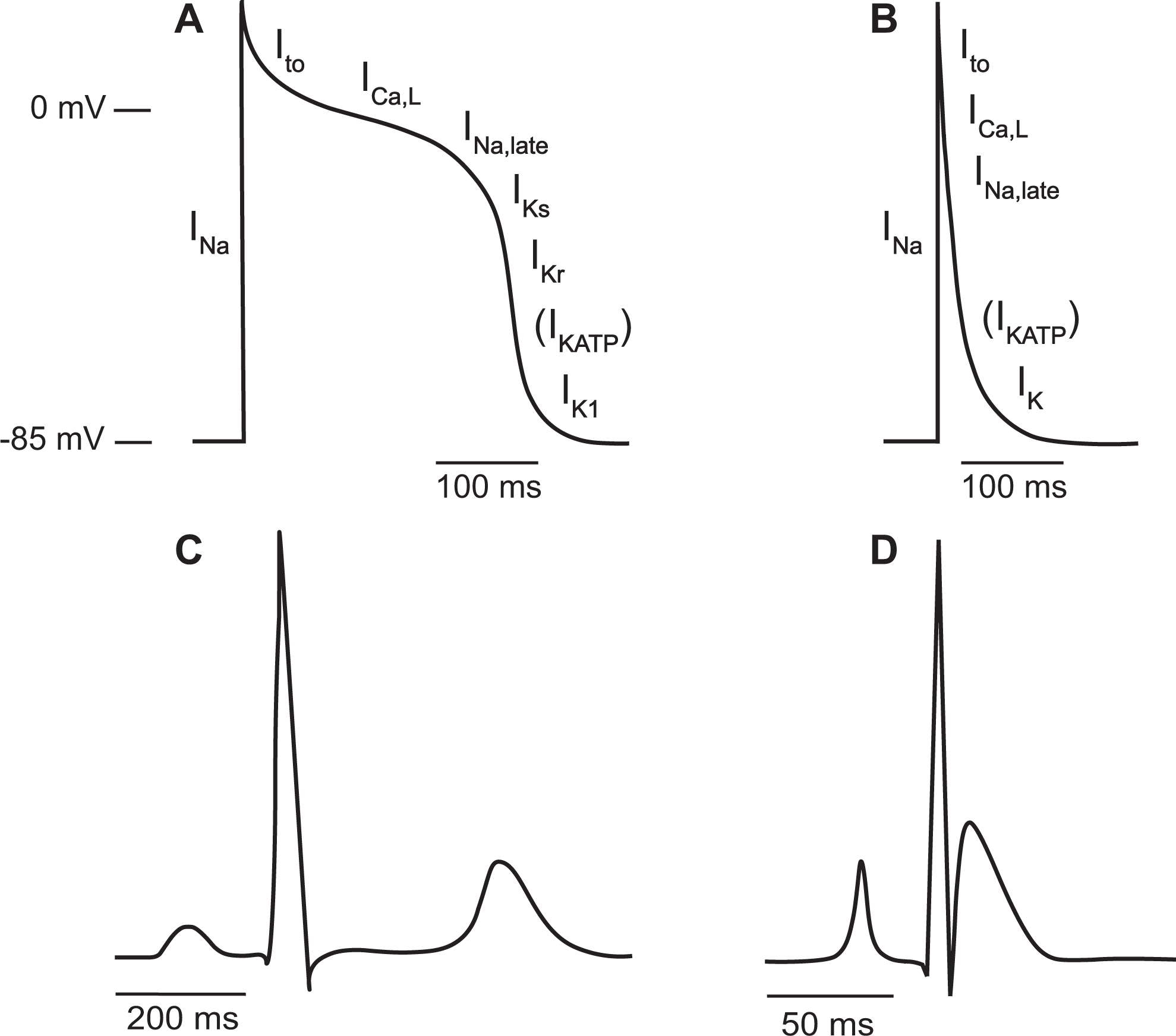
Illustration of the human (A) and rat (B) ventricular action potential (AP) and the ionic currents that govern it. While the inwards, depolarizing currents are very similar between the 2 species, the rat AP is characterized by a rapid repolarization owing to a large transient outward current (I to). The KATP channel (I KATP) does not conduct current during the normal physiological resting condition but opens upon ischemia. Illustration of the human (C) and rat (D) lead II electrocardiogram (ECG). The rat ECG distinguishes itself from the human one by lack of a distinct ST segment and a short QT interval.
Additionally, there are large anatomical differences between rat and human hearts, especially when it comes to size. The small size of the rat heart can be problematic, since it is known that a correlation between heart mass and occurrence of reentrant arrhythmias exists. 16 In addition, the heart size renders mapping studies difficult, impeding investigations on the mechanisms behind arrhythmias.
Finally, several interventions that have proven effective antiarrhythmic results in AMI rats have not yielded similar results in the clinic. For example, most investigators have found antiarrhythmic effects of class I drugs 25,34,51,53,153 -155 and class IV drugs 34,107,165 -168 in rats, whereas these compounds have no effect on SCD in humans. 8,10 -12
Practical Considerations
Other factors play a part in the choice of model as well. The rat is a very common laboratory animal for good reason. Rats are small, inexpensive, and easy to handle. The surgical procedure most often performed to produce AMI in the rat is relatively simple and does not require any highly sophisticated equipment. An alternative small animal suitable for studying arrhythmias during AMI is the mouse. Like rats, mice are inexpensive, easy to maintain and reproduce rapidly. Furthermore, mice can be genetically modified for mechanistic investigation. However, mice deviate even further from humans with respect to heart rate and size. 185 For a long time, it was questioned whether VF could occur in such a small heart mass. 191,192 Later, VF has successfully been produced in murine hearts by application of, for example, electrical pacing protocols 185 and knockouts in combination with exercise stress 193 ; however, it has been notoriously difficult to produce VF by ischemia or AMI alone. Recently, a couple of studies successfully induced VF in isolated ischemic mouse hearts by reperfusion following global ischemia, 194,195 and one group developed an ex vivo model of regional ischemia in which VF could be induced by ligation alone but only in the presence of catecholamines in the perfusate. 196 As for in vivo models, VF was evoked in a single study upon reperfusion, 197 though a number of similar studies could not induce VF, 198 -200 and one study managed to induce VF by ischemia only. 201 Yet, VF occurred in only 2 of 13 wild type (WT) mice, and again, similar studies were unable to induce VF. 197,198,200,202 This problem is absent in rats, which are often chosen for AMI studies because of high VF incidence. Thus, the literature on murine models for AMI-induced VF is quite limited compared to rats, perhaps owing to the combination of a heart size that is difficult to handle, difficulty in producing VF and scarceness of reference material.
Another reason for the wide use of rats in the study of AMI pathophysiology is their coronary anatomy. Rats have very few collateral branches, 23,203 which renders possible the production of a reproducible and consistent infarct size, opposed to, for example, dogs or guinea pigs in which the same point of ligation may result in considerable fluctuations in infarct size and arrhythmia incidence due to an extensive collateral network. 204 -206 Which model best compares to the human condition is debatable as functional collaterals are rare in young individuals but are often developed with age in particular following coronary artery disease. 207,208
Summary
Due to its acute and life-threatening nature, very little is known about the time course and the mechanisms of VF development during acute myocardial ischemia and infarction in humans, and we are yet to discover a drug which can effectively prevent it. The lack of knowledge on human ischemic VF and of an effective drug that can serve as a positive control renders model validation impossible. However, the clinical value of animal models of VF during acute myocardial ischemia and infarction can still be evaluated according to how well they recapitulate human cardiovascular physiology and pathophysiology. In this regard, the in vivo rat model possesses both advantages and disadvantages as outlined above. Though larger animal models such as dogs and pigs are closer to man in terms of cardiac anatomy and electrophysiology and though mice provide the opportunity of genetic modification, rats likewise possess advantages over other species, most importantly that they are convenient and easy to work with, and that occlusion causes predictive sequelae (including a high VF incidence), which can easily be reproduced. For this reason, the AMI rat model has played a prominent role in preclinical trials on antiarrhythmic drugs and continues to be of relevance in the development of future antiarrhythmic therapies.
Footnotes
Acknowledgments
The authors thank L. Skibsbye for providing microelectrode measurements of rat and human ventricular action potentials (unpublished data) from which we created the action potential illustrations of Figure 2.
Author Contributions
Hundahl contributed to conception and design; acquisition, analysis, and interpretation; drafting manuscript; and critically revising manuscript. Tfelt-Hansen contributed to acquisition, critically revising manuscript, and gave final approval. Jepsersen contributed to conception and design, interpretation, critically revising manuscript, and gave final approval. All authors agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by P. Carl Petersen’s Foundation Grant 16011 (to LAH) and Novo Nordisk Foundation Synergy program.