
Abstract
Background:
The development of heart failure is associated with changes in the size, shape, and structure of the heart that has a negative impact on cardiac function. These pathological changes involve excessive extracellular matrix deposition within the myocardial interstitium and myocyte hypertrophy. Alterations in fibroblast phenotype and myocyte activity are associated with reprogramming of gene transcriptional profiles that likely requires epigenetic alterations in chromatin structure. The aim of our work was to investigate the potential of a currently licensed anticancer epigenetic modifier as a treatment option for cardiac diseases associated with hypertension-induced cardiac hypertrophy and fibrosis.
Methods and Results:
The effects of DNA methylation inhibition with 5-azacytidine (5-aza) were examined in a human primary fibroblast cell line and in a spontaneously hypertensive rat (SHR) model. The results from this work allude to novel in vivo antifibrotic and antihypertrophic actions of 5-aza. Administration of the DNA methylation inhibitor significantly improved several echocardiographic parameters associated with hypertrophy and diastolic dysfunction. Myocardial collagen levels and myocyte size were reduced in 5-aza-treated SHRs. These findings are supported by beneficial in vitro effects in cardiac fibroblasts. Collagen I, collagen III, and α-smooth muscle actin were reduced in a human ventricular cardiac fibroblast cell line treated with 5-aza.
Conclusion:
These findings suggest a role for epigenetic modifications in contributing to the profibrotic and hypertrophic changes evident during disease progression. Therapeutic intervention with 5-aza demonstrated favorable effects highlighting the potential use of this epigenetic modifier as a treatment option for cardiac pathologies associated with hypertrophy and fibrosis.
Introduction
Cardiac hypertrophy, characterized by thickening of the myocardium, occurs as an adaptive response to pressure overload, metabolic disarray, mutations of sarcomeric proteins, or loss of contractile mass from prior infarction. 1 Consequently, left ventricle (LV) mass increases while chamber size may remain unchanged or decrease (eccentric hypertrophy) or increase (concentric hypertrophy), leading to impaired cardiac function. Left ventricular hypertrophy (LVH) eventually leads to congestive heart failure (HF), a major cause of morbidity and mortality worldwide. 2,3 Development of HF is associated with adverse LV remodeling and is becoming an ever more prevalent condition as risk factors such as hypertension, diabetes, and obesity continue to be poorly controlled. 4 –6
Most drivers of cardiac injury and stress cause an increase in extracellular matrix (ECM) deposition as well as myocyte hypertrophy, and it is the combination of these factors that lead to LVH and decreased organ function. Exaggerated ECM deposition or fibrosis is common in cardiac hypertrophy, and it has been suggested that alterations in the cardiac interstitium may contribute to changes in diastolic function of hypertrophied hearts. 7 It has also been suggested that myocardial fibrosis may restrict myofibrillar motion and thereby impair overall cardiac function. 8 Fibrosis is essentially an overactive wound healing response characterized by fibroblast accumulation, proliferation, and activation. In response to tissue injury, fibroblasts differentiate into contractile α-smooth muscle actin (α-SMA)-positive myofibroblasts and synthesize ECM components including collagens and fibronectin. This is an important physiological adaptation that maintains organ structural integrity after injury, but when inappropriately controlled, exaggerated myofibroblast activation can have a significant impact on the mechanical properties of myocardial tissue. The resultant remodeling of the heart ultimately leads to a decrease in ventricular compliance together with ventricular hypertrophy, conduction abnormalities, increased blood pressure, and endothelial dysfunction. 9
Physiological adaptive myocyte hypertrophy is likely predominantly driven by mechanical mechanisms. In response to pressure or volume overload, individual cardiac myocytes become mechanically stretched and activate intracellular hypertrophic signaling pathways leading to the activation of embryonic transcription factors and increased synthesis of various structural and contractile proteins.
It is likely that sustained alterations in fibroblast phenotype and myocyte activity involve reprogramming of gene transcriptional profiles and epigenetic alterations in chromatin structure. Defined as a set of mechanisms that influence gene expression without altering the DNA sequence, epigenetics plays an essential role in the regulation of gene expression. Alterations include DNA methylation, histone modifications, and micro-RNA changes.
DNA methylation involves the addition of methyl groups to cytosine residues already incorporated into DNA sequences. The reaction is catalyzed by the DNA methyltransferase (DNMT) enzymes and results in the formation of 5-methylcytosine. This generally causes gene repression either by physically preventing transcription factor binding or by reducing access through local chromatin condensation.
Interestingly, the pathogenesis of fibrosis in several other organ systems is associated with gene expression changes involving DNA methylation. In the lung, alterations in DNA methylation patterns have been associated with deregulated expression of a number of genes associated with fibroblast activation and pulmonary fibrosis. 10 –13 In addition to this, a role for DNA methylation in liver and renal fibrosis has also been alluded to. Separate studies have identified gene-specific hypermethylation events that contribute to fibroblast differentiation. 14,15
Given the associations highlighted earlier linking hypermethylation and the development of fibrosis in other organ systems together with the lack of effective therapeutics targeting the cardiac fibrotic response, we hypothesize that epigenetics may play a role in cardiac hypertrophy and fibrosis and that intervention with the DNA methylation inhibitor 5-azacytidine (5-aza) may have beneficial antifibrotic effects. 5-Azacytidine can bind to and inhibit the actions of DNMTs, the enzymes responsible for maintaining the methylation pattern of cells, resulting in a global DNA hypomethylation and subsequent alterations in gene expression. 5-Aza and 5-aza-2-deoxycytidine (5-azadC) are currently licensed for the treatment of myelodysplasic syndromes (MDSs). This 5-aza/5-azadC treatment of MDS is believed to exert its primary antineoplastic effects by directly incorporating into DNA causing hypomethylation with the resultant reexpression of epigenetically silenced genes. In the heart, drug-induced DNA hypomethylation is likely to affect many genes, and this may have a positive effect on cardiac hypertrophy and fibrosis due to the inevitable involvement of networks of proteins and pathways in reprogramming hypertrophy and fibrosis.
Therefore, the purpose of this study was to examine the effect of DNA methylation inhibition with 5-aza in a spontaneously hypertensive rat (SHR) model of cardiac hypertrophy and fibrosis and to determine whether there was a potential role for drug repurposing in the treatment of these pathologies.
Methods
Primary Cell Culture and Treatments
Primary human ventricular cardiac fibroblast (HCF) cells were purchased from ScienCell Research Laboratories, Carlsbad, CA, USA. Cells were cultured and maintained in Dulbecco modified Eagle medium (DMEM; Gibco, Paisley, UK), supplemented with 10% fetal bovine serum (Gibco) and penicillin–streptomycin antibiotics (Gibco) in a 5% CO2 humidified incubator at 37°C. When required, HCF cells were treated for up to 8 days with either 5 ng/mL human recombinant transforming growth factor β1 (TGF-β1; R&D Systems, Abingdon, UK), 5 µmol/L 5-aza (Sigma, Wicklow, Ireland), or with both simultaneously.
Rat cardiac myoblast cells (H9c2) were purchased from American Type Culture Collection (ATCC) and were maintained in low bicarbonate DMEM under the conditions described earlier. In vitro experiments were repeated n = 3 times.
Quantitative Real-Time Polymerase Chain Reaction
RNA isolation from HCF and H9c2 cells was achieved using NucleoSpin RNA II Kit (Macherey-Nagel, Duren, Germany). Total RNA was also isolated from frozen rat myocardial tissue stored in RNAlater Tissue Reagent (Qiagen, Manchester, UK). RNA extraction was performed using an RNeasy mini kit according to the manufacturer’s instructions (Qiagen). RNA quality and concentration were then determined by spectrophotometry (Nanodrop-Thermo Scientific, Hertfordshire, UK).
First strand complementary DNA (cDNA) synthesis was carried out using SuperScript II RT (Invitrogen, Paisley, UK). Quantitative real-time polymerase chain reaction (QPCR) primers were designed so that 1 of each primer pair was exon/exon boundary spanning to ensure only mature messenger RNA (mRNA) was amplified. Primer sequences are as follows: α-SMA, 5′-CGTTACTACTGCTGAGCGTGA-3′ (forward), 5′-AACGTTCATTTCCGATGGTG-3′ (reverse); collagen 1 α1 (COL1A1), 5′-GAACGCGTGTCATCCCTTGT-3′ (forward), 5′-GAACGAGGTAGTCTTTCAGCAACA-3′ (reverse); and collagen 3 α1 (COL3A1), 5′-AACACGCAAGGCTGTGAGACT-3′ (forward), 5′-GAACGAGGTAGTCTTTCAGCAACA-3′ (reverse). The QPCRs were normalized by amplifying the same cDNA with β-2-microglobulin primers, 5′-AGGCTATCCAGCGTACTCCA-3′ (forward), 5′-CCAGTCCTTGCTGAAAGACA-3′ (reverse) and myosin-binding protein C3 (MYBP-C3), 5′- CTGGAGACCTGGACCTCAGA-3′ (forward), 5′-CCGGAAACTGCTCTTCTTCA-3′ (reverse).
The QPCR was performed using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen). Amplification and detection were carried out in duplicate with Mx3000P System (Stratagene, Texas, USA). The PCR cycling program consisted of 40 three-step cycles of 15 seconds/95°C, 30 seconds/TA, and 30 seconds/72°C. To confirm signal specificity, a melting program was carried out after the PCR cycles were completed. Relative fold change in gene expression was calculated with the ΔΔCT method.
Western Blotting
Whole cell protein lysates were generated using RIPA Lysis Buffer (Millipore) with a protease inhibitor cocktail (Roche, Hertfordshire, UK). Protein concentrations were determined with a BCA Protein Assay Kit (Pierce, Hertfordshire, UK). Protein lysates were denatured, reduced, and resolved on sodium dodecyl sulfate (SDS) polyacrylamide gels by SDS-polyacrylamide gel electrophoresis before transfer onto 0.45-µm pore size Immobilon-P polyvinylidene fluoride membranes (Millipore, Cork, Ireland).
Membranes were incubated with blocking buffer (Tris-buffered saline, 0.25% Tween-20, 0.1% serum from species that secondary antibody was raised in, and 5% or 10% fat-free skimmed milk) for 1 hour at room temperature. Membranes were probed overnight with anti-α-SMA (Sigma) or anti-COL1A1 (Santa Cruz, Texas, USA) antibodies. Detection of the specific binding of the primary antibody was achieved using HRP-conjugated secondary antibodies, followed by signal detection with Immobilon Western chemiluminescent HRP substrate (Millipore). Anti-GAPDH (Millipore) was used to verify equal loading.
5-Azacytidine Treatment of a Rat Model of Hypertensive Heart Disease
Approval from the local Animal Research Ethics Committee was sought and obtained to investigate the impact of 5-aza on cardiac structure, function, and fibrosis. All animals used in the study received humane care, and the study protocol complied with the institution’s guidelines. Male SHRs and their normotensive counterparts—Wistar Kyoto (WKY) rats—were purchased from Charles River, East Lothian, UK. From 10 weeks of age, the animals received alternate day intraperitoneal injection of either phosphate-buffered saline (PBS; vehicle) or 5-aza (10 mg/kg) 14 for 12 weeks. 5-Aza (Sigma) was diluted in sterile PBS and filtered through a 0.22-µm filter. Aliquots were stored at –20°C and used within 5 days of reconstitution. The study design consisted of 3 groups of 10 animals; group 1 included 10 SHR rats which received 5-aza (SHR-5-aza), group 2 included 10 SHR rats which received PBS vehicle (SHR-V), and group 3 included 10 WKY rats which received PBS vehicle (WKY-V). All rats were housed in an animal facility under identical conditions, with a 12-hour light–dark cycle.
Systolic Blood Pressure Measurements
Systolic blood pressure was measured using the noninvasive tail-cuff method (Letica Scientific Instruments LE 5001, Barcelona, Spain). Blood pressure values were recorded while the animals were under inhaled anesthesia (2% isoflurane). The mean of 3 consecutive measurements was obtained for each animal at study midpoint (6 weeks) and end of the study (12 weeks).
Doppler Echocardiography
Cardiac structure and function was assessed at baseline and at the end of the study (12 weeks) using echocardiography. During the procedure, the animals were under inhaled anesthesia (isoflurane 2%), and body temperature was maintained using a heat mat. Echocardiography assessment was performed using a Vevo 770 High-Resolution In Vivo Micro-Imaging System (Visualsonics, Ontario, Canada) with a 10-mHz transducer. M-mode and 2-dimensional images were obtained in the parasternal long- and short-axis views. The interventricular septal thickness, posterior wall thickness, and LV diameter were measured in systole and diastole at the tips of the papillary muscle. Measurements were taken over 3 consecutive cardiac cycles and averaged. Left ventricular mass (LVM) was calculated according to Devereux formula and indexed to tibial length (LVM index [LVMI]). Blinded analysis was performed by 2 separate independent observers. Tissue Doppler images of the lateral mitral valve annulus were acquired and peak E′ determined.
Myocardial Rat Tissue Collection and Preparation
On completion of the in vivo rat study, animals were killed (terminal bleed while under inhaled 4% isoflurane anesthesia), and the heart was removed en bloc to study the impact of 5-aza on collagen deposition within the myocardium. Two methodological approaches were used to quantify collagen deposition, staining of cardiac tissue sections using picrosirius red, and hydroxyproline assay using tissue lysates.
For picrosirius red staining, the left ventricular midsections (papillary level) of the hearts were dissected immediately following killing, rinsed in PBS, and fixed with 10% formalin (Sigma). Formalin-fixed tissue was embedded in paraffin, and 5-μm thick tissue sections were created for collagen analysis using picrosirius red.
For hydroxyproline quantification, the left ventricular base of the heart was dissected immediately following killing, rinsed in PBS, and snap-frozen in liquid nitrogen until required for analysis. Frozen hearts were thawed on ice and individually disrupted and homogenized using an Ultra Turrax T25 Dispersing Instrument (IKA, Staufen, Germany). Total protein within tissue lysates were quantified, and 10 µg of homogenate was used to determine hydroxyproline content.
Hydroxyproline Assay
In brief, 500 µL of homogenized cardiac tissue sample (ratio of 100 mg tissue homogenized in 1 mL PBS) was incubated at 37°C in a vacuum oven overnight in 1-mL 6 N hydrochloric acid (HCl). Five microliters of citrate/acetate buffer (7.24% sodium acetate, 5% citric acid, 3.4% sodium hydroxide, 1.2% glacial acetic acid, and pH 6.0) and 100-μL of chloramine T-solution (282-mg chloramine T, 2-mL N-propanol, 2-mL H2O, and 16 mL citrate/acetate buffer) were added to 5 μL of the digested cardiac tissue sample and incubated for 20 minutes at room temperature. Following incubation, 100 μL of Ehrlich solution (2.5-g 4-[dimethylamino] benzaldehyde, 9.3-mL N-propanol, and 3.9-mL 70% perchloric acid) was added to each sample and incubated for 20 minutes at 65°C. Samples were subsequently cooled for 10 minutes and read at 550 nm using a SpectraMax M2 plate reader (Molecular Devices) with SoftMax Pro software (Molecular Devices, version 4.7.1). In parallel, a hydroxyproline standard curve was created to generate quantifiable data. Hydroxyproline (Sigma) concentrations from 0 to 200 μg/mL were used and were handled in a similar fashion to the digested homogenized cardiac tissue samples.
Picrosirius Red Staining and Automated Digital Quantification
Tissue sections were deparaffinized and rehydrated prior to incubating with 0.2% phosphomolybdic acid for 2 minutes. After rinsing in distilled water, the slides were stained with picrosirius red (Direct Red 80 dissolved in picric acid, Sigma) for 90 minutes. Finally, the slides were placed in 0.4% HCl for 2 minutes, 70% ethanol for 45 seconds, dehydrated, and cover-slipped for analysis.
The degree of collagen deposition was quantified by automated digital image analysis (Aperio ScanScope XT Slide Scanner; Aperio Technologies) at 20× magnification. Automated image analysis was performed using Imagescope (Aperio, Wetzlar, Germany). A positive pixel count algorithm was used to automatically quantify the area occupied by the dark pink stain colors representing collagen within each scanned slide image. Calibration of individual staining patterns was performed by specifying the requisite color (range of hues and saturation) and limits for the desired intensity range. Required input parameters for each stain were based on the hue, saturation, and intensity (HIS) color model. To detect the dark pink color of collagen with picrosirius red, a hue value of 0.8 was specified. The hue width value of 0.5 was used to allow inclusion of a moderate range of color shades. A perivascular collagen volume fraction was calculated based on the percentage of dark pink collagen staining quantified around each blood vessel and adjusted to luminal area. 16 –18
Cardiomyocytes Size Measurement
Relative cardiomyocyte size was quantified in hematoxylin and eosin (H&E)-stained mid-LV sections. Cell area was measured in transverse section by planimetry in the presence of a centrally located nucleus. Two independent observers were blinded, and 3 fields of 10 myocytes per slide (30 cells in total per section) were measured using the Aperio digital image analysis system. 19,20
Statistical Analysis
Comparisons between the control and the treatment groups were made using independent t test or analysis of variance (Tukey post hoc analysis), where appropriate, with P values <.05 considered statistically significant. All statistical calculations were performed using GraphPad prism Software (Version 4, San Diego, California).
Results
5-Azacytidine Reduces TGF-β1-Induced Collagen 1, Collagen 3, and α-SMA mRNA Expression in Human Cardiac Fibroblasts
The effect of DNA methylation inhibition on the expression of collagens 1 and 3 was examined in TGF-β1-stimulated fibroblasts by QPCR. Cardiac fibroblasts were pretreated with 5 µmol/L 5-aza for 4 days prior to stimulation with 5 ng/mL TGF-β1. An increase in expression of collagens 1 and 3 was seen in TGF β1-stimulated cells. Treatment with 5-aza significantly reduced this increase in collagen 1 (P < .01) and collagen 3 (P < .001; Figure 1A and B). A significant increase in α-SMA gene expression in TGF β1-treated fibroblasts was seen, and there was a trend toward reduction in this marker upon 5-aza treatment (Figure 1C).
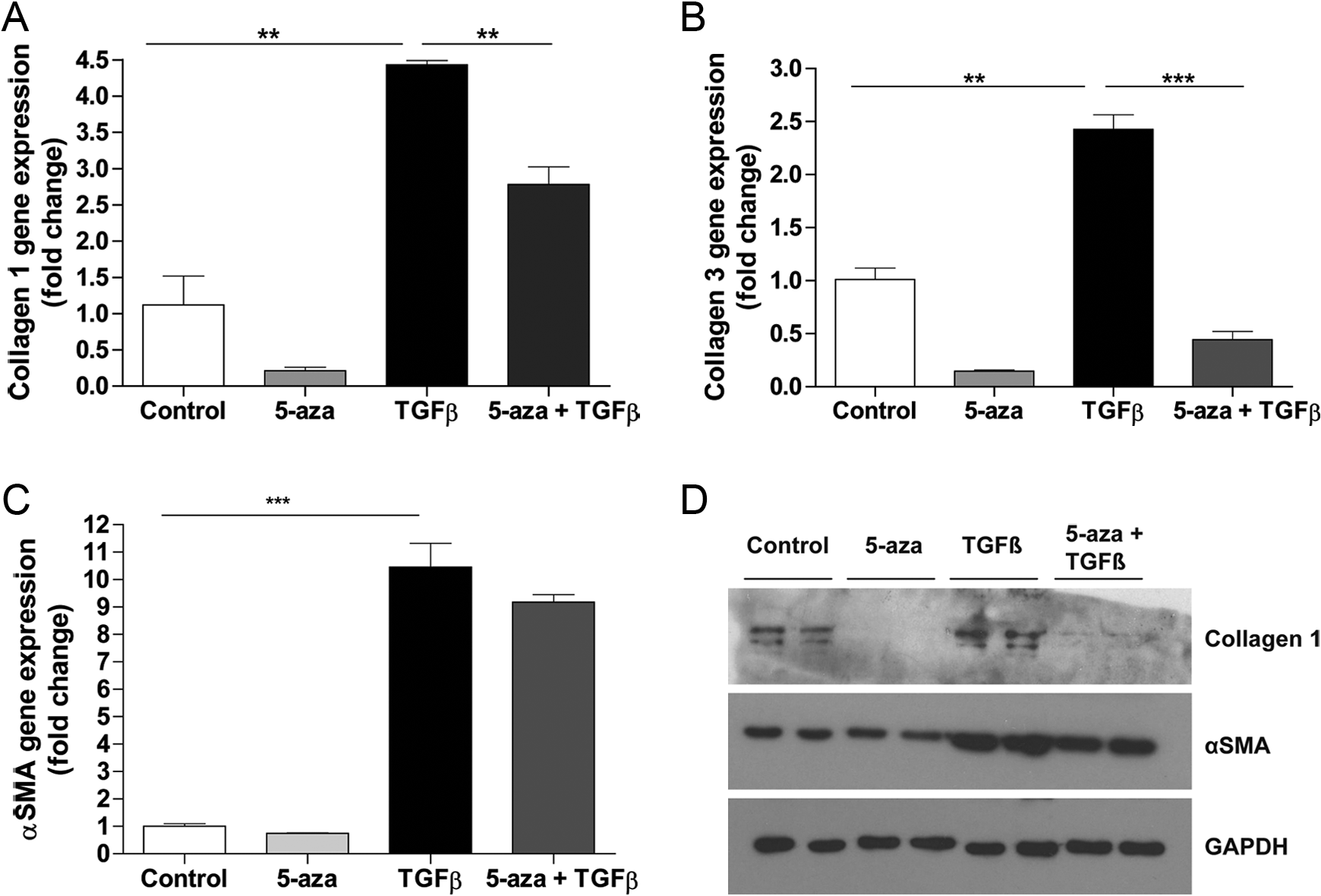
Inhibiting cardiac fibroblast DNA methylation reduces the gene and protein expression of profibrotic markers. Expression of collagen 1 α1 (COL1A1), collagen 3 α1 (COL3A1), and α smooth muscle actin (α-SMA) genes (A, B, and C) and collagen 1 and α-SMA protein (D) in human cardiac fibroblasts treated with +/– 5-µmol/L 5-azacytidine (5-aza) and +/– 5 ng/mL transforming growth factor β 1 (TGF-β1). Bars represent fold expression changes to untreated control. Data represent mean ± standard error of the mean (SEM). **P < .01, ***P < .001.
5-Aza Decreases TGF-β1-Induced Protein Expression of Collagen 1 and α-Smooth Muscle Action in Human Cardiac Fibroblasts
Collagen 1 and α-SMA protein expression were analyzed to assess the effect of 5-aza on TGF β1-induced fibroblast differentiation and activation (Figure 1D). An increase in both collagen 1 and α-SMA expression was observed in the stimulated cells. Treatment with 5-aza ameliorated TGF β1-induced collagen 1 expression. Treatment with TGF-β1 also increased α-SMA protein expression, and 5-aza significantly reduced its expression (Figure 1D).
Effect of 5-Aza on Echocardiographic Parameters and Blood Pressure
Echocardiography was performed at baseline and 12 weeks to evaluate heart structure and function. Ventricular hypertrophy was assessed using M-mode echocardiography (Figure 2A). Interventricular septum diameter (IVSD) and LVMI were used to assess LVH. The IVSD and LVMI were significantly increased in SHR compared to WKY normotensive controls. Therapeutic intervention with 5-aza significantly reduced both IVSD and LVMI at 12 weeks, P < .01 (Figure 2B and C). Diastolic function was assessed in all animals with E prime (E′) as a robust echocardiographic measure of diastolic dysfunction. The SHR group displayed a significant reduction in E′ compared to normotensive WKY control. E′ was significantly improved following 5-aza treatment (P < .05), and levels were comparable to those of normotensive WKY controls (Figure 2D). Reduced ejection fraction (EF) in the SHR group was also significantly improved with 5-aza treatment (Figure 2E). Systolic blood pressure (SBP) was assessed at 12 weeks. Blood pressure was significantly elevated in the SHR group. Administration of 5-aza had no effect on the hypertension observed in the SHR (Figure 2F).

Effects of 5-azacytidine (5-aza) treatment on echocardiographic parameters in an in vivo rat model of hypertensive heart disease. Impact of 12-week treatment of spontaneously hypertensive rats (SHRs) with 10-mg/kg 5-aza (3 doses per week) (SHR-5-aza). Normotensive Wistar Kyoto (WKY) rats and phosphate-buffered saline (PBS) vehicle-treated SHR (SHR-V) were used as control groups. Ten animals were used in each of the 3 study groups. A, Representative M-mode echocardiographic images demonstrating different degrees of wall thickness between the 3 study groups at 12 weeks. B-F, Beneficial effects of therapeutic treatment of SHR with 5-aza on cardiac hypertrophy: interventricular septal diameter (IVSD; B) and left ventricular mass index (LVMI; C); diastolic function, E′ (D); ejection fraction (EF; E), and systolic blood pressure (SBP; F). Data represent average of 2 blinded observers and graphed as mean ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001.
Cardiac Tissue Analysis
Effect of 5-Aza on Myocardial Fibrosis
The impact of 5-aza administration on collagen levels was assessed. Total soluble collagen in cardiac tissue homogenates was determined by hydroxyproline assay. A significant reduction in total collagen was observed in SHR rats treated with 5-aza compared to SHR vehicle controls, P < .05 (Figure 3A).

Effect of 5-azacytidine (5-aza) treatment on myocardial collagen deposition. Assessment of total soluble myocardial collagen (hydroxyproline assay) and perivascular collagen (tissue picrosirius red staining) within the myocardium of SHR-V and SHR-5-aza animals (A and B, respectively). C, An example of a low magnification sample image (×5 magnification) of picrosirius red-stained myocardial collagen deposition (pink staining). Both interstitial and perivascular collagen staining is apparent. A markup image is presented which was generated following the application of the positive pixel count algorithm. Regions of blue represent negative staining pixels. Yellow, orange, and red pixels represent weak, moderate, and strong positive staining collagen, respectively. Data represent mean ± standard error of the mean (SEM). *P < .05.
Histological analysis of collagen was assessed by picrosirius red staining of paraffin-embedded sections. Collagen was found to be localized to perivascular areas. The degree of collagen deposition, reflected by the magnitude of picrosirius red staining, was quantified by automated digital image analysis. A positive pixel count algorithm was used to automatically quantify the area occupied by stain colors within each scanned slide image. Application of the algorithm generated a markup image highlighting positive pixels as yellow, orange, or red and negative pixels as blue.
Results from digital analysis highlight that perivascular collagen deposition was significantly reduced in animals treated with 5-aza compared to untreated SHR animals, P < .05 (Figure 3B). Figure 3C is a representative image highlighting the creation of a markup image following the application of the positive pixel count algorithm with regions of blue representing negative staining pixels and the yellow, orange, and red pixels representing weak, moderate, and strong positive staining collagen, respectively.
Effect of 5-Aza on Cardiac Myocytes In Vivo
Myocyte area is directly related to cardiac hypertrophy. To assess the ability of 5-aza to reduce myocyte hypertrophy cell area was measured in H&E-stained cardiac tissue. Quantification using the Aperio digital image analysis system revealed that the average myocyte area in SHR-5-aza animals was significantly smaller than that of their counterpart SHR controls, P < .001 (Figure 4A). This result is consistent with the observed reduction in LVMI observed during echocardiographic analysis.

Effect of 5-azacytidine (5-aza) treatment on myocyte area in vivo and in vitro. A, Effect of 5-aza treatment of SHR animals on cardiac myocyte area obtained by digital analysis of myocyte area in hematoxylin and eosin (H&E) stained cardiac tissue sections compared to the vehicle-treated SHR animals were used as a control group. B, Gene expression changes of myosin-binding protein C3 (MYBP-C3) in SHR-5-aza versus SHR-V control animals. C, The MYBP-C3 gene expression changes in cardiac myoblast cells treated in vitro with 5-aza (4 days, 5 µmol/L). Bars represent fold expression changes to untreated control. Data represent mean ± standard error of the mean (SEM). **P < .01, ***P < .001.
5-Azacytidine Upregulates Gene Expression of MYBP-C3
Dysregulation of the cardiac MYBP-C3 gene is linked to cardiac hypertrophy, with up to 40% of hypertrophic cardiomyopathies exhibiting disease-causing mutations in this gene. 21 Upregulation of MYBP-C3 may have beneficial effects on the myocardium. 22 Inhibiting DNA methylation may also improve cardiac function through upregulation of this gene.
Treatment with 5-aza significantly upregulated MYBP-C3 gene expression in the SHR group treated with this inhibitor compared to vehicle-treated control animals (Figure 4B). In addition, myoblasts treated with 5-aza in vitro displayed a significant induction of MYBP-C3 gene expression (Figure 4C).
Discussion
Cardiac hypertrophy and fibrosis are key pathological components of the majority of cardiac diseases that are a result of myocyte hypertrophy and fibrotic collagen deposition. Therapeutic approaches for these pathologies at present target risk factors that drive the disease and do not directly modify the heart. As a result, novel approaches to treatment are urgently required.
The importance of epigenetics in cardiovascular disease is becoming ever apparent, and several studies now allude to associations between epigenetic modifications and the development of cardiac disorders. Previous reports have implicated changes in histone acetylation/deacetylation 23 –25 and differential expression of microRNAs 26 –28 in the pathogenesis of cardiac disease.
Our work has uncovered a role for DNA methylation in maintaining cardiac homeostasis. We show, for the first time, that 5-aza treatment in SHRs has beneficial effects on both myocardial fibrosis and myocyte hypertrophy. This is supported by in vitro experiments in human cardiac fibroblasts where significant protective effects of 5-aza on a profibrotic phenotype are also observed. Administration of the DNA methylation inhibitor significantly reduced gene expression levels of collagen I and collagen III in human ventricular fibroblasts. Protein analysis of collagen 1 and α-SMA, a marker of myofibroblast differentiation, also demonstrates attenuating effects of 5-aza on accumulation of these profibrotic markers.
The SHR model is characterized by ventricular hypertrophy, reactive cardiac fibrosis, and increased cardiac stiffness. 29 Echocardiographic analysis of cardiac function demonstrates favorable effects of chronic 5-aza administration. Analyzing myocardial wall dimensions is useful for the assessment of hypertrophy. Therapeutic intervention with 5-aza significantly reduced IVSD and LVMI. Left ventricular mass is one of the most accurate measures of LVH in vivo, and this is made more sensitive when indexed to body surface area in humans or tibial length in rats. 30 Myocyte hypertrophy was also assessed postmortem. Inhibiting DNA methylation with 5-aza resulted in reduced average myocyte area, thus supporting the results from the echocardiographic analyses. We hypothesize that these improvements in hypertrophy as a result of modulating DNA methylation are likely to reduce the future development of cardiovascular events and improve disease prognosis.
Furthermore, diastolic function was significantly improved. Diastolic dysfunction is the result of myocardial remodeling which can lead to abnormal LV filling, reduced LV compliance, and increased diastolic pressure, despite sustained systolic function. 31 This syndrome can eventually lead to HF with preserved EF. Diastolic dysfunction is often associated with myocardial hypertrophy and fibrosis, and some reports have shown that therapies that reduce ventricular fibrosis can improve diastolic function. 32,33 This supports our findings whereby 5-aza prevented a decrease in E′, a robust echocardiographic marker of diastolic dysfunction found to be reduced in SHRs. Administration of 5-aza maintained E′ at levels equivalent to the normotensive control group.
Postmortem tissue analysis was also undertaken. The impact of 5-aza on fibrosis and collagen deposition was examined ex vivo. Soluble collagen levels, assessed by hydroxyproline assay, were significantly reduced with DNA methylation inhibition. This finding was further supported by tissue staining analysis of perivascular collagen accumulation. Perivascular fibrosis typically precedes interstitial fibrosis and is a key determinant of adverse prognosis that impacts on myocardial oxygen delivery, tissue stiffness, and arrhythmogenic risk. 34,35 Therapeutic intervention with 5-aza significantly decreased perivascular collagen levels compared with SHR vehicle-treated animals.
An interesting finding from this study is the lack of impact of 5-aza on SBP. Blood pressure was elevated in SHR vehicle-treated animals and was unchanged in the 5-aza-treated SHR group. Given this, it is reasonable to postulate that the beneficial effects of 5-aza on hypertrophy and fibrosis are a result of modulation at the myocardial tissue level rather than as an indirect effect secondary to blood pressure reduction. Of note, while SBP remains unchanged along with a reduction in myocardial mass in the 5-aza-treated SHR group, this might have an impact on wall stress. Over time an increase in systolic wall stress could be detrimental on ventricular function. Therefore, when applying a therapeutic intervention that reduces myocardial mass in patients with high SBP, it would be important to ensure antihypertensive medications are also administered and optimized to reduce the likelihood of an increase in wall stress occurring. Taken together, the above-mentioned results highlight the importance of this epigenetic alteration in myocyte hypertrophy and myocardial fibrosis and suggest that therapeutic intervention with an epigenetic modifier such as 5-aza may be useful for treating diseases associated with these pathologies.
Our study builds on a growing body of evidence that suggests a therapeutic role for DNA methylation inhibition for the treatment of cardiovascular disorders. We have previously investigated the profibrotic impact of hypoxia on cardiac fibroblasts and examined whether alterations in DNA methylation could play a role in this process. 36 Supporting the findings that implicate a role for DNA methylation-induced fibrosis in other organ systems, we highlighted that hypoxia-induced profibrotic changes were associated with global DNA hypermethylation and increased expression of DNMT enzymes in cardiac tissue. Perhaps the most significant finding from this work was the beneficial effect upon the application of the DNA methylation inhibitor 5-azadC that inhibited the profibrotic effects of TGF β in cardiac fibroblasts. 36
Furthermore, a recent study demonstrated increased DNMT3a in activated cardiac fibroblasts and in isoprenaline-treated animals which was associated with RASSAF1A silencing and elevated α-SMA expression. Therapeutic intervention with 5-aza restored RASSAF1A expression and reduced expression of profibrotic markers. 37 A mouse model of myocardial infarction and a norepinephrine-induced model of cardiac hypertrophy also alluded to protective effects of inhibiting DNA methylation with 5-aza. 38,39 In the recent study by Xiao et al, the authors report that norepinephrine-induced cardiac hypertrophy was associated with DNA hypermethylation and an increase in cardiac levels of DNMTs. These changes were associated with alterations in the myocardium proteome as indicated using a mass-spectrometry approach. Treatment with 5-Aza during the last 6 days of norepinephrine infusion significantly reduced the associated hypertrophy, global DNA hypermethylation, and corrected the expression pattern of most of the norepinephrine-induced protein alterations in the myocardium.
The implication of epigenetics has been addressed in both HF and cardiomyopathies, where differential patterns of DNA methylation were identified in patients with these conditions. 40 –42 Movassagh et al recently uncovered a link between DNA methylation and cardiomyopathies. A genome-wide study of DNA methylation in end-stage cardiomyopathy hearts showed that the profile of this epigenetic mark differed significantly from that of healthy hearts within the CpG islands of promoters and within gene bodies. 42
Our work highlights a novel protective effect of 5-aza in a model of hypertensive heart disease which may be useful for the treatment of hypertrophic cardiomyopathy (HCM). The HCM is a disease that commonly arises due to mutations in the sarcomeric genes. It is estimated that 1 in 500 people have HCM, and cardiac MYBP-C3 mutations are reported as one of the most common genetic abnormalities. 21 A study evaluating the methylation status of MYBP-C3 revealed that the gene exhibits methylation sites and therefore exhibits potential to demethylate and increase its expression levels. 41 Reexpressing or increasing the expression levels of MYBP-C3 through gene transfer in vivo has been shown to improve systolic and diastolic contractile function as well as reduce left ventricular wall thickness. 22 Our novel data show that treatment of SHRs with the DNA methylation inhibitor 5-aza results in a significant increase in MYBP-C3 expression within the myocardium, and this coincides with reduced hypertrophy. These effects of 5-aza are confirmed in vitro in cardiac myoblast cells, whereby treatment with 5-aza upregulates MYBP-C3 gene expression. Although beyond the scope of this study, it would be of value to study the gene-specific methylation status of MYBP-C3 in disease models to provide a direct link with demethylation and improved cardiac structure and function.
In summary, although epigenetic alterations have been implicated in the development of many cancers, 43,44 the role of epigenetic changes in myocardial hypertrophy, fibrosis, and other cardiac diseases remains relatively underappreciated. Based on our findings, together with the above-mentioned evidence linking DNA methylation and cardiac dysfunction, it seems plausible to suggest that DNA methylation plays a role in the pathogenesis of cardiac disease and pharmacological modulation of this process may bring about a beneficial clinical treatment regime. Caution must be noted though regarding the need to further refine the dose and treatment regime of 5-aza therapy to minimize potential side effects. As the mechanism by which 5-aza inhibits hypertrophy and fibrosis remains unclear, it will be important to identify the affected genes in this process to facilitate the identification of novel mechanism-related therapeutic targets.
Footnotes
Author Contributions
Chris J. Watson contributed to conception and design, acquisition, analysis, and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agreed to be accountable for all aspects of work ensuring integrity and accuracy. Stephen Horgan and Roisin Neary contributed to design, acquisition, analysis, and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agreed to be accountable for all aspects of work ensuring integrity and accuracy. Isaac Tea and Niamh Corrigan contributed to acquisition and analysis, gave final approval, agreed to be accountable for all aspects of work ensuring integrity and accuracy. Nadezhda Glezeva contributed to acquisition and analysis, critically revised manuscript, gave final approval, agreed to be accountable for all aspects of work ensuring integrity and accuracy. Ken McDonald contributed to interpretation, critically revised manuscript, gave final approval, and agreed to be accountable for all aspects of work ensuring integrity and accuracy; Mark Ledwidge contributed to interpretation, drafted manuscript, critically revised manuscript, gave final approval, and agreed to be accountable for all aspects of work ensuring integrity and accuracy. John Baugh contributed to conception and design, interpretation, critically revised manuscript, gave final approval, agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Authors’ Note
Chris J. Watson and Stephen Horgan contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Authors C.W., M.L., K.M., and J.B., have a pending patent application part based on this work.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding support was provided by University College Dublin Seed Funding Scheme and the Health Research Board of Ireland.