
Abstract
Background:
Prostaglandin E2 receptor subtype 3 (EP3), a Gi protein-coupled receptor activated by prostaglandin E2, plays a particular role in cardioprotection. This study aimed to investigate the impact of EP3 deletion on cardiac remodeling and further elucidate the related involvement of possible signaling pathways.
Methods and Results:
The animals used were EP3 receptor knockout (EP3KO) mice and wild-type (WT) litter mate controls at 16-18 weeks old. The high-resolution echocardiography and weight index indicated that eccentric cardiac hypertrophy might occur in EP3KO mice, which were having worse cardiac function than WT litter mates. Isolated adult myocytes from EP3KO hearts showed spontaneous lengthening. Cardiac fibrosis was observed in EP3KO mice through Masson trichrome staining. The elevated messenger RNA (mRNA) level in matrix genes and the reduced mRNA, protein, and activity levels of matrix metalloproteinase 2 (MMP-2) indicated an increased synthesis and suppressed degradation of matrix collagen in EP3KO mice. The phosphorylation level of extracellular signal-regulated kinase (ERK) 1/2 protein was reduced in the cardiac tissue of EP3KO mice, accompanied by no significant change in the protein level of total ERK1/2, total p38, phospho-p38, glycogen synthase kinase-3β (GSK3β), phospho-GSK3β, and calcineurin (CaN) as well as CaN activity.
Conclusion:
EP3 knockout in cardiac tissues could induce eccentric cardiac hypertrophy and cardiac fibrosis at 16-18 weeks old. These effects of EP3 knockout might be regulated through inactivating MAPK/ERK pathway and affecting the MMP-2 expression. Overall, PGE2-EP3 is necessary to maintain the normal growth and development of the heart.
Introduction
In order to maintain or augment cardiac output, mature cardiac tissue adapts to increased workload or pathologic insults via growth and remodeling, including hypertrophy, fibrosis and extracellular matrix (ECM) restructuring, and necrosis and apoptosis. 1 Cardiac remodeling is a sole risk factor for cardiovascular events, such as coronary heart disease, arrhythmias, heart failure, and even sudden cardiac death. 2 In response to chronic pressure overload, concentric hypertrophy occurs, which is characterized by thickening of the ventricular wall without chamber enlargement. On the contrary, chronic volume overload often leads to eccentric hypertrophy, which is accompanied by ventricular cavity dilatation without increase or even with decrease in ventricular wall thickness. 1,3
Prostaglandin E2 (PGE2), an important lipid metabolite, is derived from arachidonic acid by cyclooxygenases (COXs) and PGE synthases (PGES). Prostaglandin E2 plays a crucial role in an array of physiological and pathophysiological functions, including the regulation of blood pressure, cell proliferation, and inflammatory response. 4 Prostaglandin E2 is a potent inflammatory mediator, and its production can be reduced by nonsteroidal anti-inflammatory drugs (NSAIDs) through pharmacological inhibition of COXs. Nonsteroidal anti-inflammatory drugs are classified into 2 types based on their selective inhibition of 2 different COX isoforms. One type is traditional NSAIDs (tNSAIDs), such as aspirin and ibuprofen, which inhibit both COX isoforms at standard doses. Another type is selective COX-2 inhibitors (coxibs), which are widely used in the clinic due to their superior gastrointestinal safety compared to nonselective tNSAIDs. However, long-term use of coxibs increases the risk of serious cardiovascular events, including myocardial infarction, congestive heart failure, and sudden cardiac death. 5,6 This fact raises a special concern for the relationship of coxibs and heart diseases. Prostaglandin E2 is considered an important downstream effector of COXs, and thus, they and their receptors become research focus for understanding the pathogenic mechanisms of cardiovascular complications upon coxibs administration.
Prostaglandin E2 interacts with 4 distinct G protein-coupled membrane receptors EP1 through EP4, which differ in terms of signaling pathways involved. 7 Prostaglandin E2 receptor subtype 1 couples to Gq-proteins to increase intracellular calcium; EP2 and EP4 receptors couple to Gs-proteins and increase intracellular Cyclic adenosine monophosphate (cAMP) concentration, whereas EP3 mainly couples to Gi-proteins to decrease intracellular cAMP concentration and mobilize intracellular calcium. It follows that different EP receptors have distinct or even opposite functions. In modulation of blood pressure homeostasis, for example, EP1 and EP3 receptors mediate the pressor response, whereas EP2 and EP4 receptors mediate the depressor response. 8
Prostaglandin E2 plays a particular role in protecting the reperfused myocardium from ischemic injury via EP3 9 and EP4 10 receptors. Prostaglandin E2 binding to EP4 receptor contributes to normal and adaptive growth of cardiomyocytes. 10 -14 In contrast, EP3 overexpression attenuates myocardial injury during ischemia and reperfusion 9 and also activates the prohypertrophic calcineurin (CaN) signaling pathway. 15 And EP3 inactivation attenuates angiotensin II pressor response via decreasing arterial contractility, however, no alleviated cardiac hypertrophy was observed. 16 So far, the relationship between EP3 expression and cardiac hypertrophy has not yet been fully studied.
In this study, we aimed to determine whether cardiac hypertrophy and dysfunction were induced in mice with a global targeted disruption of EP3 receptor and further investigate the possible involvement of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway.
Methods
Experimental Animals
The global EP3 receptor knockout (EP3KO) mice (male, 16 to 18 weeks old) and sex- and age-matched wild-type (WT) litter mates (serving as controls) on a pure C57BL/6 background were kindly gifted by Dr Richard M. Breyer, Vanderbilt University School of Medicine. The study protocols and the use of the animals were reviewed and approved by the Animal Care and Use Review Committee of Peking University Health Science Center. The study conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no 85-23, revised 2011).
Genotyping of EP3KO Mice
Genotyping of WT and EP3 mutant alleles was performed using polymerase chain reaction (PCR)-based strategies. After weaning, tail DNA was isolated for this section. Two pairs of specific primers (EP3+/+ and EP3−/− in Table 1) were used to amplify a 152 and 721 base pair (bp) genomic DNA fragment, sparing the mutant site for WT and EP3KO mice, respectively. Polymerase chain reactions were carried out for both WT and EP3 alleles. The conditions included denaturation at 94°C for 5 minutes, then 94°C for 30 seconds, 58°C for 30 seconds, and 72°C for 30 seconds for 35 cycles, followed by a final extension at 72°C for 6 minutes. Polymerase chain reaction products were separated on a 1% agarose gel.
Sequence of Primers for Genotyping and RT-PCR Analysis.

Abbreviations: bp, base pair; COX, cyclooxygenase; EP, prostaglandin E2 receptor subtype; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MMP, matrix metalloproteinase; mPGES1, microsomal prostaglandin E synthase-1; RT-PCR, reverse-transcriptase polymerase chain reaction; TIMP, issue inhibitors of metalloproteinase.
Echocardiography
Twenty-two mice (16 to 18 weeks old) were used for this study (11 WT and 11 EP3KO). All animals were anesthetized by injection of 2% pentobarbital sodium into the abdominal cavity (10 mg/100 g body weight), then placed on a heating pad. Echocardiograms were performed for cardiac imaging using a Vevo 2100 system with a MS400C scan head (Visual Sonics, Toronto, Ontario, Canada). The left ventricular (LV) wall dimensions and fractional shortening (FS) were calculated from parasternal imaging of the short axis of the heart at the papillary muscle level. Imaging at the long axis was acquired to calculate the ejection fraction (EF), the ratio of mitral valve early diastolic to atrial contraction flow velocity (MV E/A), and LV volume and mass. Measurements were taken from 5 consecutive cardiac cycles, and calculations were performed using built-in formulas of the Visual Sonics cardiac measurements package. 17 The echocardiography was performed by the same investigator who was blinded to the genotype.
Isolation of Cardiac Myocytes
Sixteen- to 18-week-old mice were anticoagulated with heparin (5 IU/g) and anesthetized with pentobarbital (10 mg/100 g body weight). Hearts were removed and arrested in diastole via cannulation for retrograde perfusion by the method of Langendorff 18 with Ca-free perfusion buffer (NaCl 140 mmol/L, KCl 5.4 mmol/L, NaH2PO4·2H2O 1.8 mmol/L, MgCl2·6H2O 1.2 mmol/L, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 5 mmol/L, taurine 20 mmol/L, glucose 10 mmol/L; pH 7.30 with NaOH and gassed with 95% O2:5% CO2). Ventricular cardiac myocytes were isolated by collagenase II (Worthington, New Jersey) digestion followed by a brief mechanical disassociation. Images of adult myocytes were obtained under fluorescent-inverted microscope. Myocyte length and width were determined using ImageJ software. Approximately 80 cells were evaluated per mouse heart.
Cardiac Tissue Sampling
After echocardiography, hearts were removed from animals for weighing, and then the ratio of heart weight to body weight (HW/BW) was calculated. The cardiac tissue samples were kept in liquid nitrogen at −80°C for subsequent analysis.
Histological Analysis
Another set of cardiac tissue samples was fixed with 4% paraformaldehyde and cut into 5-μm thick paraffin sections within the similar ventricular regions, which were subjected to Masson trichrome staining. The collagen fibers were stained blue, the nuclei were stained black, and background was stained red. Five different visual fields per section were chosen randomly and photographed under the 20× objective lens. Interstitial collagen fraction (ICF) was measured using QWin software package (Leica, Bensheim, Germany) avoiding perivascular collagen and averaged across all 5 fields of the sections.
RNA Extraction and Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was extracted from cardiac tissue samples using TRIpure reagent (BioTeke, Beijing, China) as described by the supplier, and complementary DNA (cDNA) was synthesized from 2 μg of total RNA with RevertAid first strand cDNA synthesis kit (Fermentas, Burlington, Ontario, Canada) according to the kit instructions. Quantitative real-time reverse-transcriptase polymerase chain reaction (RT-PCR; qPCR) was performed with the SYBR Green I probe (Bio-Rad, Hercules, California). The PCR protocol was denaturation at 95°C for 10 minutes, then 95°C for 15 seconds, and 60°C for 1 minute for 40 cycles, followed by a final extension at 72°C for 6 minutes. The primer sequences are listed in Table 1. Each sample was analyzed in triplicate and normalized to the level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) messenger RNA (mRNA). The 2−ΔCt method was used to evaluate the mRNA level in tissue samples from EP3KO mice and WT controls.
Western Blot Analysis
The cardiac tissue sample (approximately 50 mg) was mechanically disrupted in radioimmunoprecipitation assay lysis buffer (Beyotime, China) containing Phenylmethanesulfonyl fluoride (PMSF). A total of 40 μg of protein per sample was resolved in 12% denaturing polyacrylamide gels (Bio-Rad). Extracellular signal-regulated kinase 1/2, p38 MAP kinase (p38), glycogen synthase kinase 3β (GSK3β), and CaN, along with matrix metalloproteinase 2 (MMP-2) were detected with the appropriate antibodies: antibodies against ERK1/2 or p-ERK1/2 (1:500), antibodies against p38 or p-Thr180/Tyr182 p38(1:1000), and antibodies against GSK3β or p-Ser9 GSK3β (1:1000) from Cell Signaling Technology (Massachusetts); anti-CaN (1:500) from Abcam (Cambridge, United Kingdom); and anti-MMP-2 polyclonal antibody (1:500) from Santa Cruz Biotech (Texas). Anti-GAPDH polyclonal antibody (1:800; Santa Cruz Biotech) served as control for normalization. The antibody–antigen reaction was detected using a horseradish peroxidase-conjugated secondary antibody (1:2000; Jackson ImmunoResearch, Pennsylvania). Immunoreactivity was visualized by enhanced chemiluminescence (Cell Signaling). Band intensities were quantified with Quantity One software (Bio-Rad).
Gelatin Zymography
Matrix metalloproteinase 2 activity of heart tissue was determined by gelatin zymography. Protein samples (50 μg) were mixed with loading buffer lacking reducing agents (such as β-mercaptoethanol) and subjected without boiling to 10% sodium dodecyl sulfate/polyacrylamide gel incorporating 1 mg/mL of gelatin B (Sigma-Aldrich, Germany). After electrophoresis, the gel was washed in 2.5% Triton X-100 for 30 minutes at room temperature and incubated overnight (16-18 hours) at 37°C with 50 mmol/L Tris, 0.2 mol/L NaCl, 10 mmol/L CaCl2, pH 7.4. Stain the gel by 0.1% Coomassie blue R-250, 45% methanol, 10% acetic acid, and destain the gel in deionized water for 1 hour at room temperature under gentle agitation. Gel was scanned to the computer, and quantitative analysis was performed with Quantity One software (Bio-Rad).
Calcineurin (PP-2B) Activity Assay
Calcineurin activity was measured with a cellular activity assay kit (Calbiochem, Germany). After lysed in lysis buffer with protease inhibitor, tissue homogenate was centrifuged at 150 000g at 4°C for 45 minutes to get the high-speed supernatant (HSS). Excess phosphate (PO4) and nucleotides were removed from HSS extracts by gel filtration. Assays were executed in 96-well plate following the manufacturer’s protocol. The absorbance at 620 nm was measured on microplate spectrophotometer. The mean of A620,total − A620,ethylene glycol tetraacetic acid (EGTA) and A620,OA − A620,(OA+EGTA) was converted into phosphate release (nmol PO4) based on a standard curve prepared in each assay. The CaN activity was measured by phosphate release divided by total protein concentration (nmol PO4/mg protein).
Statistical Analysis
Prism (GraphPad, Software Inc, La Jolla, California) was used for statistical analysis. All data are expressed as means ± standard error of the mean. Data between 2 groups were evaluated by 2-tailed Student t tests. A value of P < .05 was considered statistically significant. Data illustrated are representative of at least 3 independent experiments.
Results
The Expression of Enzymes for Synthesis of PGE2 and the EP Receptors
The primers distinguished the WT and EP3KO mice by amplifying a 152-bp fragment in EP3+/+ gene and a 721-bp fragment in EP3−/− (Figure 1A). Quantitative PCR data indicated that the key enzymes in PGE2 biosynthesis, including COX1/2 and microsomal PGES1, along with all of the PGE2 receptors could be detected in cardiac tissues of WT mice. Since deletion of EP3 receptor may lead to a compensatory change in other EP receptors, 12 we performed RT-PCR to amplify all 4 EP receptor mRNAs. As the results shown, the mRNA level of EP3 and its subtype (EP3α, β, and γ) were significantly lower in EP3 knockout mice than in WT controls, but no differences were found in the mRNA level of other EP receptors between the 2 animal groups (Figure 1B and C), indicating that other EP receptors were not changed to compensate for the global loss of EP3.

A, Polymerase chain reaction (PCR) genotyping of wild-type (WT) and EP3 receptor knockout (EP3KO) mice. Using tail DNA, WT mice only represent a 152-base pair (bp) PCR product, whereas homozygous EP3KO mice only have a 721-bp product. B, A representative agarose gel of PCR products for EP3 and its subtype messenger RNA (mRNA) levels from hearts of WT and EP3KO mice. C, The mRNA levels of EP receptors, cyclooxygenase (COX) 1/2, and microsomal prostaglandin E synthase-1 (mPGES1) from WT (black bars) and EP3KO (open bars) hearts as determined by quantitative real-time PCR. n = 5, *P < .05, **P < .01, ***P < .001, WT versus knockout (KO).
The Changes in Cardiac Structure and Ventricular Function
To evaluate cardiac structure and ventricular function, high-resolution echocardiography was performed (Figure 2A). The measurement data are summarized in Table 2. The LV mass and corrected LV mass of EP3KO mice were significantly higher in EP3KO mice than in WT controls. This result was consistent with the difference in the ratio of HW–BW between the 2 animal groups. The LV internal diameter and volume in end-diastole and end-systolic were significantly increased in EP3KO mice as compared with in WT controls, while wall dimensions calculated from short-axis M-mode views of the LV were comparable in the 2 animal groups, indicating the structural difference between the 2 animal groups. The EF and FS, 2 parameters that measure the pumping function of the heart, both with MV E/A, which evaluates diastole function of the left ventricle, were severely depressed in EP3KO mice compared to WT controls. In addition, qPCR revealed that the mRNA level of natriuretic peptide B, skeletal muscle alpha 1 actin (Acta1), and Acta2 (the 3 markers of cardiac disease) in cardiac tissues was significantly higher in EP3KO mice than in WT controls (Figure 2B).
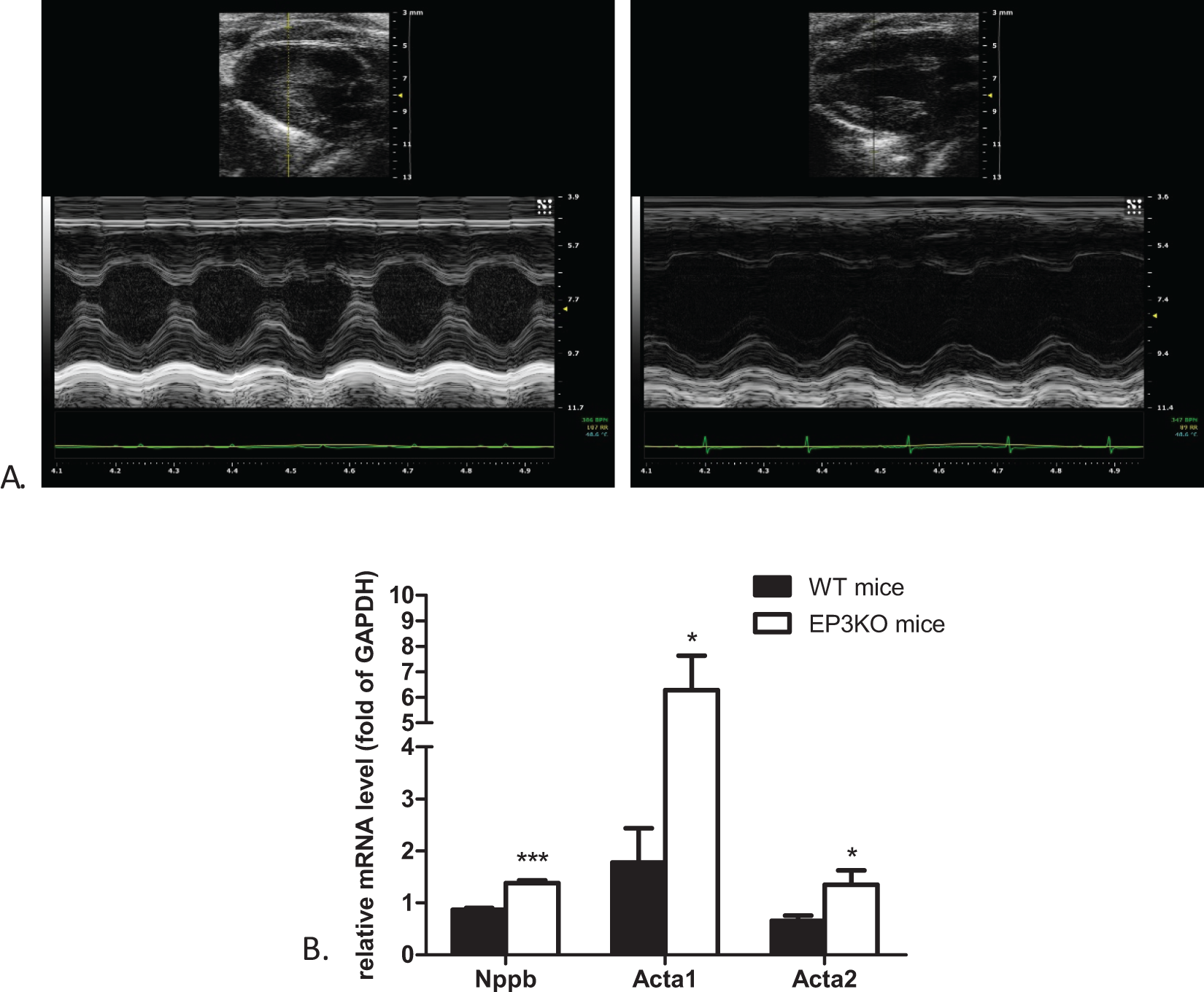
A, Representative long axis M-mode echocardiograms taken from the left ventricles of wild-type (WT; left panel) and EP3 receptor knockout (EP3KO) mice. B, Quantitative real-time polymerase chain reaction (PCR) for the hypertrophic markers Nppb, Acta1, and Acta2 in both groups of mice (*P < .05 and ***P < .001, WT vs EP3KO mice, n = 6). Nppb indicates the protein coding gene of natriuretic peptide B; Acta1 and Acta2, the protein-coding genes of α-skeletal actin.
Baseline Characteristics of WT and EP3KO Mice.a

Abbreviations: BW, body weight; EF, ejection fraction; EP3KO, EP3 receptor knockout; FS, fractional shortening; HR, heart rate; HW/BW, the ratio of heart weight to body weight; LV, left ventricle; corrected LV mass: 0.8×LV mass; LVAWd, left ventricle anterior wall thickness in diastole; LVAWs, left ventricle anterior wall thickness in systole; LVEDV, left ventricle end-diastolic volume; LVESV, left ventricle end-systolic volume; LVPWd, left ventricle posterior wall thickness in diastole; LVPWs, left ventricle posterior wall thickness in systole; MV E/A, the ratio of mitral valve early diastolic (E) to atrial contraction (A) flow velocity; WT, wild type.
an = 11.
b P < .01.
c P < .05 and for WT vs EP3KO mice.
d P < 0.001.
Morphology of Isolated Ventricular Myocyte
To understand cellular morphology, we measured the length and width of isolated myocytes from WT and EP3KO hearts (Figure 3A). Myocytes from EP3KO hearts showed a greater increase in cell length, whereas myocyte width has no difference compared with WT hearts. Furthermore, length–width ratios were more pronounced, suggesting that myocytes from hearts of EP3KO mice were significantly longer (Figure 3B).

A, Representative images of isolated adult cardiomyocytes from hearts of wild-type (WT; left panel) and EP3 receptor knockout (EP3KO) mice. B, Quantitative analysis of isolated myocyte length, width, and length/width ratio (*P < .05 and ***P < .001, WT vs EP3KO mice, n = 80 cells from 4 mice each).
Myocardial ECM Remodeling in EP3KO Mice
Masson trichrome staining (Figure 4A) was used to evaluate the degree of cardiac fibrosis in animal hearts. As compared with WT controls, EP3KO mice showed a higher ICF value, which means an increased level of interstitial fibrosis in their hearts (Figure 4B).

A, Masson trichrome staining (×20) of cross section of myocardium was used to observe the cardiac fibrosis of wild-type (WT; left panel) and EP3 receptor knockout (EP3KO) mice. B, Interstitial collagen fraction (ICF) as a percentage of area of heart analyzed in WT and EP3KO mice. n = 4, **P < .01 for WT vs EP3KO mice. C, Collagen types I and III both with the collagen-binding proteoglycans biglycan and decorin messenger RNA (mRNA) levels were measured by quantitative real-time polymerase chain reaction (PCR) with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as endogenous control. n = 6, **P < 0.01 for WT vs EP3KO mice. D, Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) mRNA levels, including MMP-2, MMP-9, MMP-14, TIMP-1, and TIMP-2, were determined by quantitative RT-PCR, and the results were normalized by GAPDH. n = 4, **P < .01 for WT vs EP3KO mice. E, ProMMP-2 and active MMP-2 in protein extracts from WT and EP3KO hearts and densitometric evaluation as shown at the right panel. The GAPDH signals indicated protein loading. n = 3, **P < .01 for WT versus EP3KO mice. F, The mature form of 62-kDa MMP-2 activity was detected by zymography, and the intensity of bands was represented at the right bar graph. n = 6, **P < .01 for WT versus EP3KO mice.
The EP3KO mice demonstrated an increased mRNA level of the heart matrix genes, including collagen I, collagen III, and collagen-binding proteoglycan biglycan, 19 compared to WT hearts (Figure 4C). In addition, the MMP-2 expression at the mRNA and protein levels in the cardiac tissue was significantly reduced in EP3KO mice compared to WT controls, but there was no difference in the mRNA level of tissue inhibitors of metalloproteinase (TIMP)-1, TIMP-2, MMP-9, and MMP-14 between the 2 animal groups (Figure 4D and E). Besides, MMP-2 activity assessed by gelatin zymography was shown to decrease in EP3KO hearts.
Signaling Pathway Analysis
We performed Western blot analysis to determine signaling pathways involved in the effect of EP3 deletion on cardiac hypertrophy. 20 The phospho-ERK1/2 protein level in the cardiac tissue of EP3KO mice was significantly decreased compared to that of WT controls, whereas no significant difference was detected in the total ERK1/2 protein level between the 2 animal groups (Figure 5A). In contrast, there was no significant difference found in the protein level of total p38, phospho-p38, GSK3β, phospho-GSK3β, or CaN (Figure 5B–D). Meanwhile, the CaN activity showed no significant difference between EP3KO mice and WT controls, as measured by a specific assay (Figure 5D).
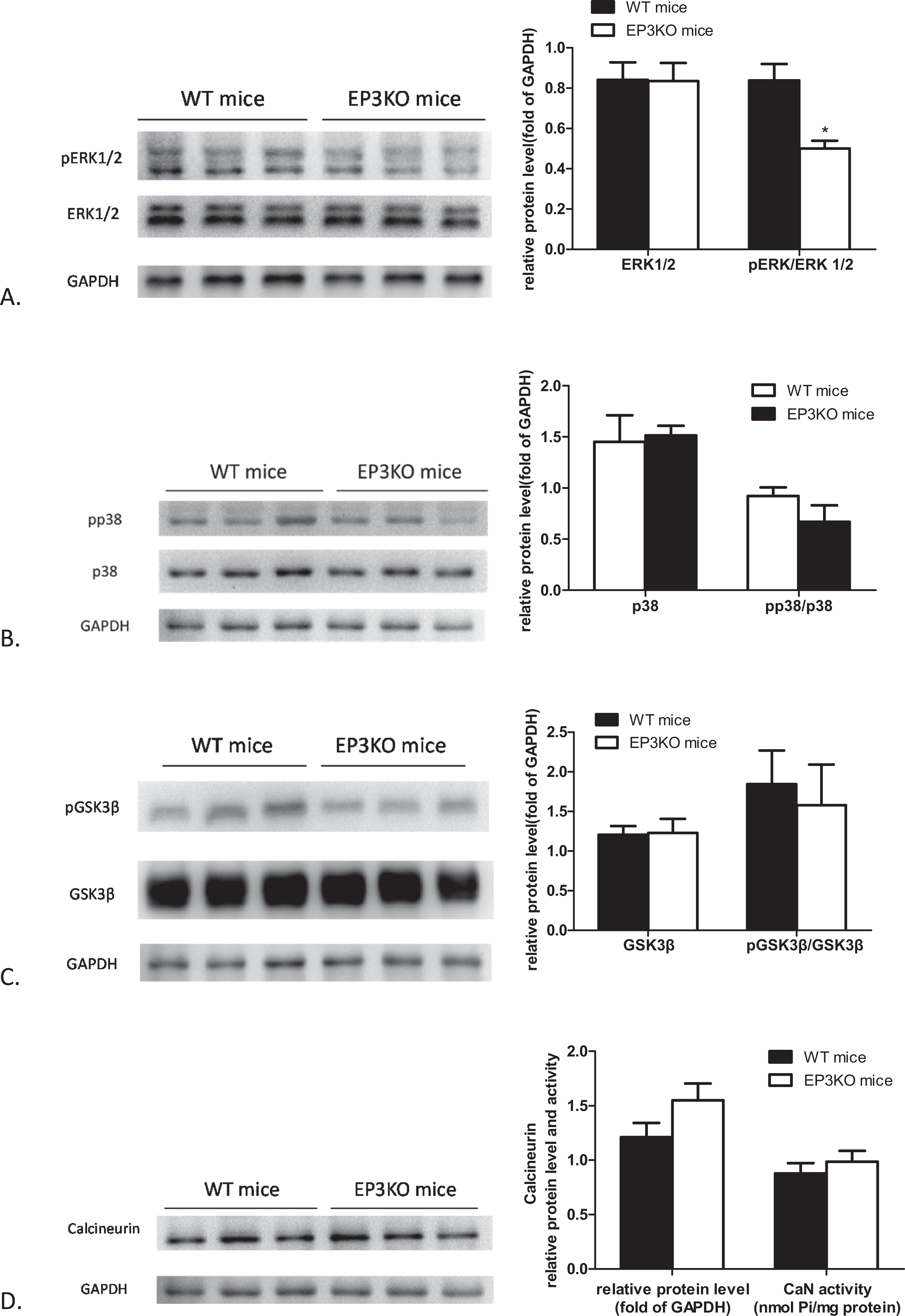
Post-EP3 signaling cascades that regulate cardiomyocyte hypertrophy. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) signals indicate protein loading. A, Total extracellular signal-regulated kinase (ERK) 1/2 and phosphorylated ERK1/2 (pERK1/2) in protein extracts from wild-type (WT) and EP3 receptor knockout (EP3KO) hearts. Right panel is the densitometric evaluation of ERK1/2 and pERK1/2. B, Total p38 and phosphorylated p38 (pp38) in protein extracts from WT and EP3KO hearts. Right panel is the densitometric evaluation of p38 and pp38. C, Glycogen synthase kinase 3β (GSK3β) and phosphorylated GSK3β (pGSK3β) in protein extracts from WT and EP3KO hearts. Right panel is the densitometric evaluation of GSK3β and pGSK3β. D, Calcineurin in protein extracts from WT and EP3KO hearts. Right panel is densitometric evaluation of calcineurin and calcineurin-phosphatase activity. n = 6, *P < .05 for WT versus EP3KO mice.
Discussion
Prostaglandin E2 is possibly the most extensively studied prostaglandin in the cardiovascular system conceivably due to its multiple receptor subtypes. 21 Several studies have investigated the importance of PGE2 to cardiac function through a relative deficiency of mPGES-1-catalyzed PGE2 biosynthesis. 22 -24 The direct effects of 4 EP receptors on cardiac function, however, remain unresolved. Overexpression of cardiac-specific human EP3-II receptor seems to serve a dual function: anti-ischemic effects and mediation of cardiomyocyte hypertrophy. 9,15 In this study, we observed that EP3 gene disruption could induce cardiac remodeling, that is, eccentric cardiac hypertrophy and cardiac fibrosis, implying a key role of EP3 receptor in maintaining cardiac function.
Earlier work from our laboratory has demonstrated that there was no noted difference between EP3KO mice and WT mice in heart rate level, which was recorded via computerized tail-cuff method in conscious state. 16 The anesthetic agents may produce low heart rate in the small animals, then lead to misleading data on cardiac parameters. 25 In this study, the difference of heart rate between these 2 groups in the anesthetized state was not statistically significant either. It could be considered that the depth of anesthesia on the measured values has been eliminated. Compared to WT litter mates, EP3KO mice had cardiac hypertrophy, as evidenced by a significant increase in LV mass and HW/BW. Echocardiography revealed a dramatic increase in left ventricular internal diamenter in diastole (LVIDd), left ventricular internal diamenter in systolic (LVIDs), left ventricle end-diastolic volume, and left ventricle end-systolic volume of EP3KO mice, but no change in wall thickness. Valve pathological changes were not observed according to echocardiography, and there was no abnormality in atrioventricular (AV) conduction through cardiac monitor in the hearts of EP3KO mice at this age. Eccentric hypertrophy is characterized by dilatation of the LV chamber, whereas concentric hypertrophy is associated with increased LV wall thickness. 26 All the data demonstrated that the heart of EP3KO mice developed an eccentric hypertrophy. This observation was different from dilated cardiomyopathy in EP4 knockout mice. 11 Although EP3KO mice had an average EF of 65.28%, a value still within the normal range, EF, FS, and MV E/A of EP3KO mice were significantly lower in comparison to those of WT litter mates. Besides, there are dozens of factors that affect the cardiac function, such as strains and age. We also observed that EF of the 2 groups decreased at 50 weeks, but the level of EP3KO mice is more notable than that of the WT control group (data not shown). Whether the EP3KO mice would develop reduced wall thickness and cardiac dilation with age needs further investigation. In addition, transcriptional changes of the cardiac fetal genes, such as B-type natriuretic peptide (BNP) and α-skeletal actin, were often seen in pathological remodeling and heart failure. Combining echocardiography data with the finding on the higher level of cardiac-specific gene transcripts in EP3KO mice, we would like to suggest that EP3 deletion enhances ventricular remodeling and impairs cardiac function.
Myocardium is composed of cardiac myocytes, nonmyocytes, and ECM. Isolated adult myocytes from EP3KO hearts showed spontaneous lengthening, which matched the morphologic change characteristic of myocytes in eccentric hypertrophy. As the highest cell population in the myocardium, cardiac fibroblasts are able to produce ECM proteins (collagens, proteoglycans, and so forth) and the ECM regulatory proteins, including MMPs and their inhibitors, TIMPs. 27 A hallmark feature of ventricular remodeling is excessive deposition of ECM to constitute cardiac fibrosis. 28 The pathological foundation of cardiac fibrosis includes the increased secretion and decreased degradation of collagen, which leads to ECM sediment. In this study, the upregulated mRNA levels of collagen type I and III in cardiac tissues of EP3KO mice suggested an increase in collagen synthesis. Biglycan, a small leucine-rich proteoglycan, is capable of specific binding to collagen types I, II, III, and VI and inhibiting collagen breakdown. 19 The mRNA level of biglycan in cardiac tissues was increased in EP3KO mice. Matrix metalloproteinases are the predominant proteases responsible for ECM protein degradation. A well-controlled dynamic balance between MMPs and TIMPs is critical for maintaining ECM homeostasis. 27 Matrix metalloproteinase 2 is secreted as inactive proproteins (proMMP-2). It has been well established that proMMP-2 can be activated with TIMP-2 and MMP-14. 29 There was no difference in the mRNA level of TIMP-2 and MMP-14 in the cardiac tissue between the 2 animal groups, indicated that the ability of proMMP-2 activation might not be affected by deletion of EP3 receptor. However, the mRNA, protein, and activity levels of MMP-2 were significantly reduced in EP3KO heart. Matrix metalloproteinase 9 and TIMP-1 are also consistently indicted in cardiovascular disease development and prognosis. 30 Yet, EP3KO mice did not display significant differences in mRNA levels of MMP-9 and TIMP-1 when compared with WT mice. Taken together, the increased biglycan level and decreased active MMP-2 level were related to suppression of collagen degradation. Both elevated synthesis and inhibited degradation of ECM contributed to the formation of cardiac fibrosis in EP3KO mice. On one hand, ventricular dilation could result from myocardial cell elongation. On the other hand, the accumulation of ECM could result in the unchanged posterior wall thickness, especially in the earliest stages of eccentric hypertrophy.
Prostaglandin E2 receptor subtype 3 signals are primarily involved in inhibition of adenylyl cyclase via Gi activation and in Ca2+ mobilization through Gβγ from Gi. 31 To further investigate the possible mechanism underlying the above observations, we determined the expression and activity of ERK1/2, p38, GSK3β, and CaN to test the involvement of MAPK/ERK, p38 MAPK, Phosphoinositide 3-kinase (PI3K)-AKT/PKB-GSK3β, and Ca2+-CaN pathways, respectively.
Phosphorylated and activated ERK1/2 is capable of phosphorylating numerous transcription factors that induce the reprogramming of cardiac gene expression. Nevertheless, blockade or deletion of cardiac ERK1/2 showed no reduction in pathologic or physiologic stimulus-induced hypertrophy in vivo. 32 Decreased or absent ERK1/2 signaling induces myocyte lengthening and eccentric hypertrophic growth. Extracellular signal-regulated kinase 1/2 is believed to be involved in regulating the balance between eccentric and concentric growth of the heart. 33 On the other hand, activating MMP-2 secretion requires the MAPK/ERK pathway, and NSAIDs could suppress MMP-2 expression by inhibiting ERK activation. 34 Our results confirmed that EP3 ablation results in an obvious decline in phosphorylation of ERK1/2. Diminished ERK1/2 activation could partially explain the eccentric growth of myocyte and the reduced expression of MMP-2 in EP3KO hearts.
p38 is another member of MAPK family. Activation of p38 in the mouse heart induces dilation and failure but not hypertrophy. However, targeted inhibition of p38 signaling induces and enhances cardiac hypertrophy by augmenting CaN–nuclear factor of activated T cells (NFAT) signaling. 20 Calcineurin, a Ca2+-activated phosphatase, mediates a hypertrophic response through dephosphorylating NFAT transcription factors in the cytoplasm, then causing the nuclear localization of NFATs. 35 As an important downstream target of AKT/PKB in the heart, GSK3β is normally active and inhibits a number of hypertrophic transcriptional effectors, and it can be inhibited by AKT/PKB-mediated phosphorylation, thereby promoting both protein synthesis and gene transcription. 36 Nevertheless, in EP3KO hearts, there was no significant change in phosphorylated p38 or GSK3β as well as CaN activation. p38 MAPK, PI3K-AKT/PKB-GSK3β, and Ca2+-CaN pathways might not be involved in cardiac remodeling and functional deficits induced by EP3 deletion. Cardio-specific overexpression of the porcine homologue of the human EP3-II receptor also could induce LV hypertrophy. 15 But, the type of cardiac growth and molecular mechanism might differ from each other.
Interestingly, reduced MMP-2 expression and activity seem to be responsible for ECM remolding in diabetic myocardiopathy. 37,38 Protein expression of MMP-2 was significantly diminished in diabetic rats, whereas MMP-9, TIMP-1, and TIMP-2 were unchanged. 39 And EP3 receptor-deficient mice exhibited a high-serum level of insulin at 3 months of age, whereas impaired glucose tolerance and insulin resistance grow with age. 40 Therefore, the occurrence and development of diabetes due to the lack of EP3 receptor may also participate in regulating cardiac MMP-2 expression and activity in EP3KO mice, although this speculation required further investigation.
Consistent with these previous findings, here, we observed ERK1/2 inactivation in EP3KO mice that had developed eccentric hypertrophic growth of hearts, suggesting that PGE2-EP3-ERK is essential to maintain the normal growth and development of the heart. Prostaglandin E2 receptor subtype 3 is unique in that it has multiple isoforms generated by alternative mRNA splicing, and these receptor isoforms display differences in regulation of signaling molecules. For example, PGE2 could strongly stimulate the phosphorylation of ERK1/2 by human EP3-II and EP3-III isoforms through different mechanisms, while ERK1/2 phosphorylation by EP3-Ia isoform was minimal. 41 Therefore, there may also exist similarities and differences among contributions of these isoforms to cardiomyopathy.
In conclusion, this study elucidated for the first time that inactivation of EP3 receptor in vivo is sufficient to induce unfavorable influence on cardiac structure and function. We also showed that MAPK/ERK pathway but neither PI3K-AKT/PKB-GSK3β pathway nor Ca2+-CaN pathway might be involved in EP3 disruption-induced eccentric hypertrophy and cardiac fibrosis through affecting the MMP-2 expression. Since PGE2-EP3 receptor prevents the heart from eccentric hypertrophy growth, the therapeutic effect of prostaglandin synthesis inhibitors, such as NSAIDs, must be properly evaluated. Besides, roles of each EP3 isoform in heart disease have not been studied yet. For further studies, it would be interesting to determine whether the functions of EP3 isoforms are different from each other, thus developing a potential therapeutic target for treating cardiac eccentric hypertrophy via selective regulation of the function of EP3 receptor.
Footnotes
Author Contributions
Shuang Liu contributed to design, acquisition, and analysis, contributed to interpretation, drafted the manuscript, and critically revised the manuscript. Jianhua Zhu, Youfei Guan, and Richard Breyer contributed to conception and critically revised the manuscript. Hongzhuan Sheng contributed to conception and design and critically revised the manuscript. Yawei Ji and Hu Xu contributed to acquisition and analysis and critically revised the manuscript. Jian Yao and Xiaodan Zhao contributed to acquisition and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (30971223, 30971224, and 81270275), Research and Innovation Project for College Graduates of Jiangsu Province (CXZZ13_0875), and Scientific and Technological Innovation Project for College Graduates of Nantong University (YKC13071).