
Abstract
Background:
With the rise of the burden of ischemic heart disease, both clinical and economic evidence show a desperate need to protect the heart against myocardium ischemia-reperfusion injury-related complications following cardiac surgery or percutaneous coronary intervention. However, there is no effective intervention for myocardium ischemia-reperfusion injury as yet.
Methods:
We pretreated mice with 4 daily 2.0 absolute atmosphere (ATA) hyperbaric oxygen, then observed its effects on heart function parameters and infarct size following in situ ischemia-reperfusion. Multiple oxidative and inflammation products were measured in the myocardium. Next, we investigated the expression of heme oxygenase 1 (HO-1), phosphatidylinositol 3-kinase (PI3K)/serine/threonine protein kinase (Akt) pathway, and NF-E2-related factor 2 (Nrf2) in the presence of myocardium ischemia-reperfusion injury, hyperbaric oxygen preconditioning, and their inhibitors and their effects on heart function parameters.
Results:
Hyperbaric oxygen preconditioning ameliorated the cardiac function and histological alterations induced by myocardium ischemia-reperfusion injury, decreased oxidative products and proinflammatory cytokine. Hyperbaric oxygen preconditioning increased expression of HO-1, which was suppressed by PI3K inhibitor LY294002, Nrf2 knockout, and Akt inhibitor triciribine. The expression of Nrf2 was enhanced by hyperbaric oxygen preconditioning, but decreased by LY294002 and triciribine. The Akt was also activated by hyperbaric oxygen preconditioning but suppressed by LY294002. The hemodynamic assays showed that cardiac function was suppressed by LY294002, Nrf2 knockout, and triciribine.
Conclusion:
These data present a novel signaling mechanism by which hyperbaric oxygen preconditioning protects myocardium ischemia-reperfusion injury via PI3K/Akt/Nrf2-dependent antioxidant defensive system.
Keywords
Introduction
Acute myocardial infarction (AMI) remains a major cause of morbidity and mortality throughout the world. Currently, early reperfusion by thrombolysis and percutaneous coronary intervention (PCI) are most effective and important therapies. 1 Numerous evidence has shown that blood flow restoration is necessary to save endangered myocardium after AMI. However, it can also induce reperfusion injury by triggering a cytotoxic cascade with overflow of reactive oxygen species (ROS), paradoxically causing contractile dysfunction, cell death, and inflammation, which is called myocardium ischemia-reperfusion injury (MIRI). 2,3 The burst of ROS overwhelms the antioxidative capacity in body, leading to cellular necrosis and apoptosis, increased myocardial infarct size, and reduced myocardial function. 2 In a cardiac surgery, this usually occurs when removing the aortic cross-clamp; in the primary PCI, this occurs when the balloon is deflated after inflation. It may induce either acute consequences such as low cardiac output and death or chronic results including heart failure. 4 It was estimated that MIRI-related complications consume US$2 billion to US health care resources each year. 5 With the rise of the burden of ischemic heart disease, both clinical 6 and economic evidence 7 show a desperate need to protect the heart against MIRI-related complications following cardiac surgery or PCI. However, there is no effective intervention for MIRI as yet, although many studies have explored potential therapeutic strategies.
Hyperbaric oxygen preconditioning (HBO-PC) is a pretreatment with hyperbaric oxygen to provide protection against a subsequent oxidative injury. 8 Accumulating evidence has shown that HBO-PC could protect against various ROS-related injuries, including focal cerebral ischemia, 9 hyperoxic seizures, 10 spinal cord ischemic injury, 11 traumatic, and surgical brain injury. 12,13 It was first reported that pretreating mice with hyperoxia (95% O2, 1 ATA) attenuated MIRI. 14 Infarct size in isolated hearts was reduced from 45% to 22% in treated animals. In 2005, Cabigas et al treated mice with 2.0 ATA HBO and found out that the myocardial infarct size in isolated heart following ischemia-reperfusion (I/R) was decreased, in which nitric oxide synthase (NOS) plays an essential role. 15 So Yogaratnam et al suggested that HBO-PC may be used to protect the myocardium from the effects of ischemia-reperfusion injury via nitric oxide. 16 However, in situ I/R model was seldom used to study the effects of HBO-PC on MIRI and the mechanism was not yet clear. Recent research suggests that the effects of HBO-PC may involve heme oxygenase 1 (HO-1) and NF-E2–related factor 2 (Nrf2) besides nitric oxide. 11,17 Hence, the present work was carried out to explore the protective effect of HBO-PC against MIRI and the underlying mechanism. We pretreated mice with 4 daily 2.0 ATA HBO, then observed its effects on heart function parameters and infarct size following in situ I/R. Multiple oxidative products in the myocardium were measured to evaluate the antioxidative effect of HBO-PC. As the inflammatory response and cytokines play an important role in the pathophysiological response to MIRI, 18 we also examined the myeloperoxidase (MPO) activity, proinflammatory cytokine tumor necrosis factor α (TNF-α), and interleukin 1β (IL-1β) levels.
Next, we investigated the role of HO-1, phosphatidylinositol 3-kinase (PI3K)/Akt pathway, and Nrf2 in the protective effects of HBO-PC to further explore the underlying mechanism. Heme oxygenase 1 is a rate-limiting enzyme with potent antioxidant, anti-inflammatory/antiapoptotic characters. 19 The HO-1 is generally accepted as an antioxidant against oxidative injury which could inhibit ROS production. It was reported that activation of an ERK/Nrf2/HO-1 signaling pathway protected cells from oxidative stress, 20 and the upregulated expression of HO-1 could ameliorate cardiopulmonary bypass-induced oxidative damage. 21 Numerous research has revealed the role of HO-1 in the protective effect of HBO-PC. The mechanism of the induction of HO-1 by HBO-PC is not clear yet. It is well known that HO-1 may be regulated by Nrf2, an important sensor of oxidative stress which maintains the redox homeostasis by regulating transcriptional activation of phase II defense and antioxidant genes, 22,23 whose activation contributes to cytoprotection induced by HBO-PC. 11 Many studies have demonstrated that PI3K/Akt pathway may be involved in the activation of Nrf2/antioxidant response element (ARE) pathway. 24 –26 Their expression and effects were examined in the study.
Materials and Methods
Animals and Reagents
Adult male wild-type and Nrf2−/− mice (C57B/SV129), which were previously described by Xu et al, 27 were bought from Shanghai experiment animal center and used in this study. The study was approved by the institutional animal care and use committee in the Fourth Affiliated Hospital animal center (Harbin Medical University) and carried out in accordance with the Guide for the Care and Use of Laboratory Animals, Eighth Edition (National Academies Press, Washington, DC, 2010). The animals were all housed in the Fourth Affiliated Hospital animal center with a 22°C temperature, 41% relative humidity, and 12-/12-hour light/dark cycles in the animal care wing and were allowed access to water and food ad libitum. Zinc protoporphyrin IX (ZnPP-IX, inhibitor of HO-1), LY294002 (inhibitor of PI3K pathway), and triciribine (inhibitor of Akt pathway) were purchased from Sigma-Aldrich (St. Louis, Missouri). ZnPP-IX (3 mg/kg of body weight), LY294002 (100 mg/kg of body weight), or triciribine (2 mg/kg of body weight) was administered intraperitoneally daily during HBO-PC exposure.
Experimental Protocol
For functional, biomarkers and histological studies, 120 wild-type animals were randomly assigned to the following 4 groups: Sham, Control, HBO-PC, and hyperbaric air-preconditioning (HBA-PC) (30 animals per group). In each group, 8 mice were used for functional studies, 6 for infarct size measurement, 8 for biomarkers measurement, and 8 for histological studies. For Western blot study, 6 mice were used in each group. The Nrf2 knockout (Nrf2-KO) mice were used in the Nrf2-KO group. In the Sham group, mice received myocardium I/R surgical procedures except that the left anterior descending artery was not occluded. In the Control group, mice received 4 daily normobaric air pretreatment, rested for 24 hours, then received myocardium I/R surgical procedures. In the HBO-PC group, mice received 4 daily HBO (2.0 ATA) pretreatment, rested for 24 hours, then received myocardium I/R surgical procedures. In the HBA-PC group, mice received 4 daily hyperbaric air (2.0 ATA) pretreatment, rested for 24 hours, then received myocardium I/R surgical procedures.
HBO-PC and HBA-PC Treatment
The HBO-PC or HBA-PC treatment was started 5 days before myocardium I/R surgical procedures. The chamber was first ventilated with 100% O2 or air at a flow rate of 2.0 L/min to minimize CO2 accumulation. Then the pressure in the chamber was increased at a rate of 1.0 ATA/min to 2.0 ATA and maintained for 1 hour. After the exposure was done, mice were then decompressed at a rate of 1.0 ATA/min and freed from the chamber.
In Situ Model of Myocardial I/R
We used the murine model of in situ myocardial I/R injury previously described by Hampton et al. 28 Briefly, mice were first intraperitoneally anesthetized with pentobarbital sodium (100 mg/kg, Abbott Laboratories, Chicago, IL, USA). Anesthesia was confirmed by lack of foot withdrawal reflex. Temperature was monitored and maintained at 37°C with a heating lamp. Then the mice were intubated and ventilated with a small animal ventilator (MiniVent, Type 845; Hugo Sachs Elektronik, March-Hugstetten, Germany). Next, a middle cervical incision was made, and a section of PE-90 tubing was passed through the exposed trachea until the tip was 2 mm below the larynx. Under a dissecting microscope, the left parasternotomy was performed by dividing 3 ribs in a cephalocaudal direction parallel to the sternum. The pericardium was opened. After the left coronary artery was located, a silk black braided suture (7-0) was inserted around the artery near its origin. A snare was created by passing both ends of the suture through the tip of a 22-gauge angiocatheter that could then be tightened and released by sliding a Voss clip down on the angiocatheter. After 60 minutes of ischemia, the occlusive snare was released for reperfusion. Sham mice underwent the same surgical procedures except that the suture was not snared. When the reperfusion was finished, for hemodynamic measurements, the muscle layer and the skin were closed and the animals were allowed to recover for 24 days. Buprenorphine hydrochloride was injected (0.65 mg/kg, intramuscularly) to reduce postoperative pain. For biomarker analyses (performed on the same day of hemodynamic measurements), the LV was dissected free, rinsed in saline, and stored at −80°C. For tetrazolium chloride (TTC; performed after the 2-hour reperfusion) and hematoxylin-eosin (H&E) studies (performed on the same day of hemodynamic measurements), hearts were rapidly harvested and cut into slices.
Measurements of Cardiac Function
Measurements of cardiac function were adapted from Li et al’s study. 29 Mice were intraperitoneally anesthetized with chloral hydrate (300 mg/kg) 24 days after the I/R treatment. A small incision was made to the right of the midline in the neck. The external right carotid artery was exposed and a microtip pressure transducer catheter (model SPR-612, Millar Instruments, Houston, Texas) was introduced into the artery. The proximal end of the catheter was connected to an electrostatic chart recorder (model ES 2000, Gould, Cleveland, Ohio). After arterial blood pressure was monitored, the inserted tip of this catheter was advanced down until it reached the left ventricular lumen. The left ventricular pressure (LVP) signal was continuously obtained, which was used to calculate left ventricular systolic pressure (LVSP), left ventricular developed pressure (LVDP), and ± (dP/dt) max.
Infarct Size Determination
After the 2-hour reperfusion, the suture around the coronary artery was retied before 2% Evans blue dye was infused into the aortic root to mark the area at risk (AAR, blue dye negative). Infarct size was determined with TTC staining. Briefly, the hearts were collected and kept at −20°C for 30 minutes. Next, frozen hearts were sectioned parallel to the atrioventricular groove in 1-mm sections, which were thawed and incubated in a 1% TTC phosphate-buffered solution (pH 7.4) at 37°C for 15 minutes. Then they were rinsed in phosphate-buffered saline. Next, they were fixed in 10% formalin to increase the contrast of the Evan’s blue and TTC staining. Subsequently, the slices were placed on a light table and photographed on both sides. The pictures were then analyzed, and the different areas were delineated. The infarct size was calculated as a percentage volume of the infarct area (white area) versus the AAR (nonblue area).
Hematoxylin-Eosin Staining
For H&E staining, the hearts were harvested and flushed with normal saline, transferred to 4% formaldehyde for 48 hours, and embedded in paraffin. The paraffin was then cut into 4-mm thick serial sections. Next, butterfly shaped sections of 4-μm thickness were cut and stained with H&E staining for histopathological analysis. Briefly, slices were deparaffinized with xylene, then rehydrated in absolute alcohol. After washing in distilled water, they were stained in hematoxylin solution for 8 minutes. Next, the slices were washed in running tap water for 5 minutes and differentiated in 1% acid alcohol for 30 seconds. After they were washed again with tap water and saturated lithium carbonate solution, they were rinsed in 95% alcohol and counterstained in eosin–phloxine solution. Finally, the slices were dehydrated again with alcohol and mounted with xylene-based mounting medium, and then assessed for the following: cardiomyocyte hydropic changes, neutrophilic infiltrate, hemorrhage, lymphohistiocytic infiltrate, and acute myocardial necrosis.
Measurement of Malondialdehyde, Protein Carbonyl, and 8-Hydroxy-2-Deoxyguanosine in Myocardium Tissues
Transmural tissue from AAR (100 mg, wet weight) was homogenized in 2 mL of 10 mmol/L phosphate buffer (pH 7.4). After centrifugation at 10 000g for 30 minutes, the malondialdehyde (MDA), protein carbonyl, and 8-hydroxy-2-deoxyguanosine (8-OHdG) content in the supernatant were measured using the corresponding kits. The protein concentration was determined using a standard bicinchonininc acid (BCA) protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China). For MDA assay, similar method to Weng et al 30 was adopted, using a commercial kit (Nanjing Jiancheng, Nanjing, China). Results were expressed as μmol/mg protein. Protein carbonyls were measured with a commercial quantitative assay kit (Cayman Chemical Company, Ann Arbor, Michigan, USA). Results were expressed as nmol/mg protein. For 8-OHdG assay, DNA was extracted from the tissue with a DNA Extraction Kit (DNA Extractor Wb Kit, Wako Chemical, Japan), then added to plate wells precoated with anti-8-OHdG antibody (Fukuroi, Japan) and incubated for 45 minutes at 37°C. The wells were washed for 3 times and sequentially treated with immunoglobulin G (IgG), streptavidin-horseradish peroxidase, and 3,3′,5,5′-tetramethylbenzidine. The incubation lasted for 15 minutes and was terminated by sulfuric acid. Finally, the absorbance was read at a wavelength of 450 nm. Results were expressed as pg/g protein.
Measurement of Myeloperoxidase, Tumor Necrosis Factor α, and Interleukin 1β Levels
Transmural tissue from AAR were harvested and washed in normal saline, and then homogenized in saline (25 mg/mL) on ice. The homogenates were centrifuged at 3000g at 4°C for 15 minutes. The levels of MPO, TNF-α, and IL-1β were then measured with an enzyme-linked immunosorbent assay kit, according to the manufacturer manuals (Sigma, St. Louis, Missouri). The levels were calculated with the absorbance read on a microplate reader. The protein concentration was determined using a standard BCA protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China), and the results were expressed as per microgram of protein.
Western Blot
Cardiac tissues were harvested and washed in ice-cold saline, and then homogenized in radioimmunoprecipitation assay buffer lysis buffer (25 mg/mL) with 1 mmol/L Phenylmethanesulfonyl fluoride (PMSF) on ice. All the samples were centrifuged at 3000g at 4°C for 15 minutes and the supernatants were collected. The protein concentration was determined using a standard BCA protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China). Protein sample of 50 mg was loaded per lane, separated on 12% dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred to nitrocellulose membranes electrophoretically. Membranes were blocked in 5% nonfat milk in TBST (10 mmol/L Tris, 150 mmol/L NaCl, 0.05% Tween-20) for 1 hour at room temperature and then blocked with first antibodies (mouse anti-HO-1, rabbit anti-Nrf2, mouse anti-phosphorylated Akt, and Akt, StressGen Biotech, California) in 5 mL of 5% bovine serum albumin wash buffer at 4°C overnight. The membrane was washed with wash buffer and incubated with horseradish peroxidase-conjugated secondary antibody anti-rabbit IgG (1:2000) in TBST solution for 1 hour. Detection was performed using the SuperSignal West Pico system (Pierce, Illinois) and exposed to X-ray film (Fujifilm, Japan).
Statistical Analysis
The data are expressed as mean ± standard deviation (SD). Statistical analysis was done using the SPSS 17.0 with 1-way analysis of variance followed by Student-Newman-Keuls post hoc test; P < .05 was considered significant.
Results
Cardiac Function Changes by HBO-PC
As shown in Figure 1, there were significant changes caused by MIRI and HBO-PC on all the hemodynamic parameters. The MIRI treatment significantly decreased ±(dP/dt) max and LVSP and increased LVDP (P < .05 compared to Sham), which were reversed by HBO-PC (P < .05 compared to Control). For ±(dP/dt) max and LVSP, the values of HBO-PC were still lower than Sham (P < .05 compared to Sham); but for LVDP, there was no significant difference between HBO-PC and Sham (P > .05 compared to Sham). The HBA-PC treatment, however, had no ability to reverse the hemodynamic changes caused by MIRI (P > .05 compared to Control).

The changes in cardiac function by myocardial ischemia-reperfusion (I/R), hyperbaric oxygen preconditioning (HBO-PC) and HBA-PC. ± (dP/dt) max, left ventricular developed pressure (LVDP), and left ventricular systolic pressure (LVSP) were measured 24 days after the I/R procedure. A and B, Changes in ± (dP/dt) max. C and D, Changes in LVDP and LVSP, respectively. Sham: mice received myocardium I/R surgical procedures except that the left anterior descending artery was not occluded. Control: mice received 4 daily normobaric air pretreatment before myocardium I/R surgery. HBO-PC: mice received 4 daily HBO (2.0 ATA) pretreatment before myocardium I/R surgery. HBA-PC: mice received 4 daily hyperbaric air (2.0 ATA) retreatment before myocardium I/R surgery. Results are expressed as mean ± standard deviation (SD; *P < .05 compared to Sham and P < .05 compared to Control, n = 8).
Infarct Size and Histopathological Changes
Figure 2A and B shows the infarction percentage of AAR and histopathological changes in each group. As shown in Figure 2A, the infarction percentage in the HBO-PC group (21% ± 1.8%) was significantly lower than that in the Control group (44% ± 2.1%; P < .05), but the difference between the HBA-PC group (38 ± 1.7%) and the Control group was not significant (P > .05). Blinded histological analysis of H&E staining results showed that the HBO-PC group had lower degrees of necrosis, hemorrhage, neutrophilic infiltrate, and spindle-shaped interstitial cells compared with the Control group. No improvement of these abnormalities was shown in the HBA-PC group.
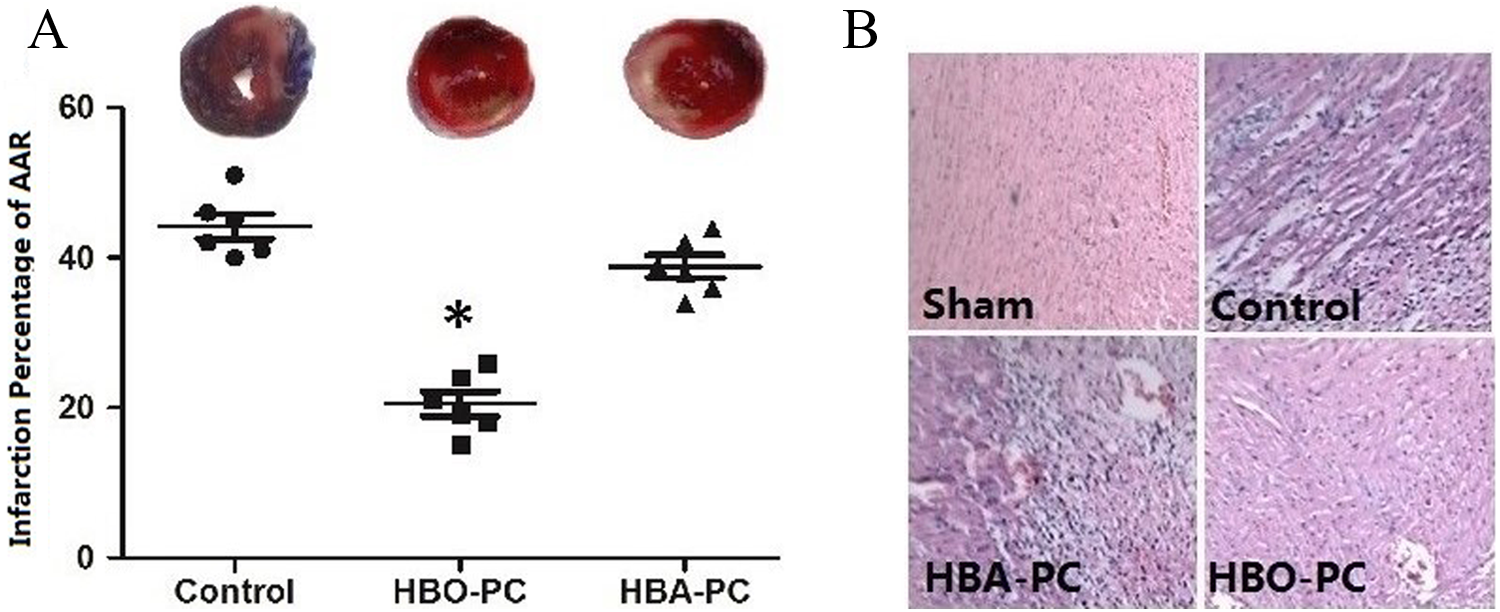
The changes in infraction size and histology by myocardial ischemia-reperfusion, hyperbaric oxygen preconditioning (HBO-PC), and HBAPC. The infraction size was measured by Evan’s blue and tetrazolium chloride (TTC) staining and presented as infraction percentage of area at risk (AAR) after the 2-hour reperfusion (A). Results are expressed as mean ± standard deviation (SD; *P < .05 compared to Control, n = 6). The histopathological changes were assessed by hematoxylin-eosin (H&E) staining on the same day of cardiac functional measurement (B). Cardiomyocyte hydropic changes, neutrophilic infiltrate, hemorrhage, lymphohistiocytic infiltrate, and acute myocardial necrosis were examined in the H&E staining (n = 8).
Changes in the Levels of Protein Carbonyl, 8-Hydroxy-2-Deoxyguanosine, and Malondialdehyde
As shown in Figure 3, myocardial MDA, protein carbonyl, and 8-OHdG assays demonstrated that MIRI caused significant oxidative stress (Sham group vs Control group, P < .05). The HBO-PC treatment significantly mitigated the oxidative stress, as shown by the dramatic decreases in the MDA, protein carbonyl, and 8-OHdG levels compared to the Control group (P < .05). The HBA-PC group showed no significant reduction of these oxidative parameter levels (P > .05 vs Control group).
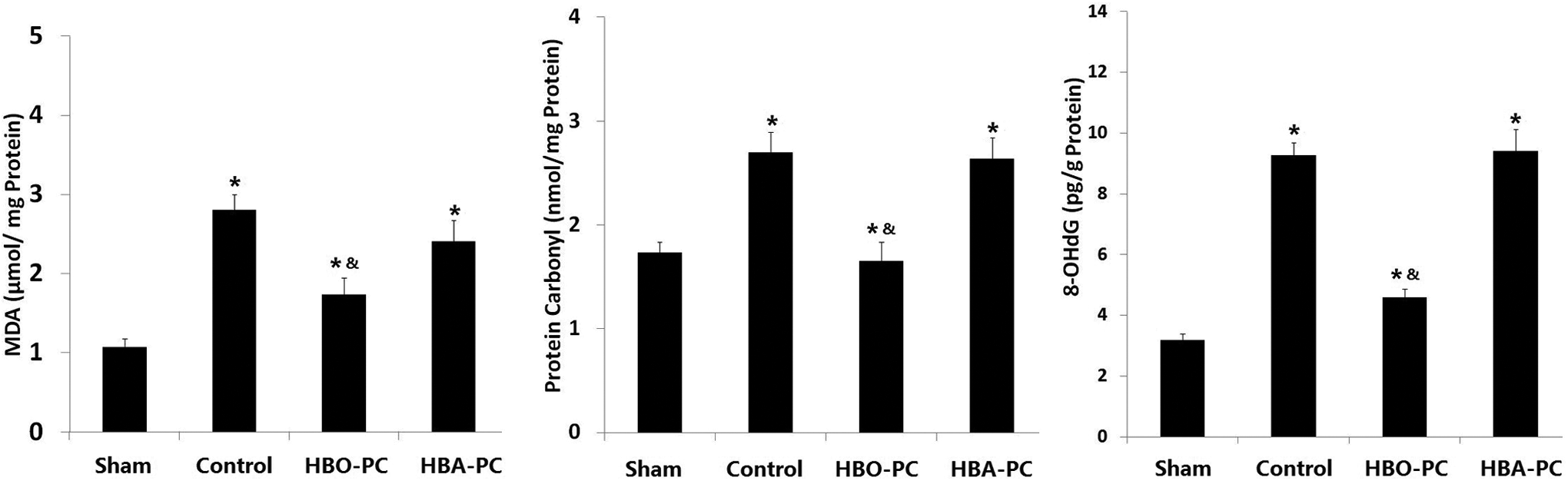
Protein carbonyl, 8-hydroxy-2-deoxyguanosine (8-OHdG), and malondialdehyde (MDA) in myocardium tissues. Myocardium oxidative injury was measured using protein carbonyl, 8-OHdG, and MDA assays 24 days after the ischemia-reperfusion (I/R) procedure. Values are the mean ± standard deviation (SD). *P < .05 compared to the Sham group and P < .05 compared to the Control group; n = 8 per group.
Changes in the Levels of Myeloperoxidase, Tumor Necrosis Factor α, and Interleukin 1β
As shown in Figure 4, MIRI caused significant increase in myocardial MPO, TNF-α, and IL-1β assays compared to the Sham group (P < .05), which was reversed by HBO-PC treatment, as shown by the dramatic decreases in the MPO, TNF-α, and IL-1β levels compared to the Control group (P < .05). The HBA-PC group, however, had no such ability as the HBO-PC group (P > .05, HBO-PC vs Control group).
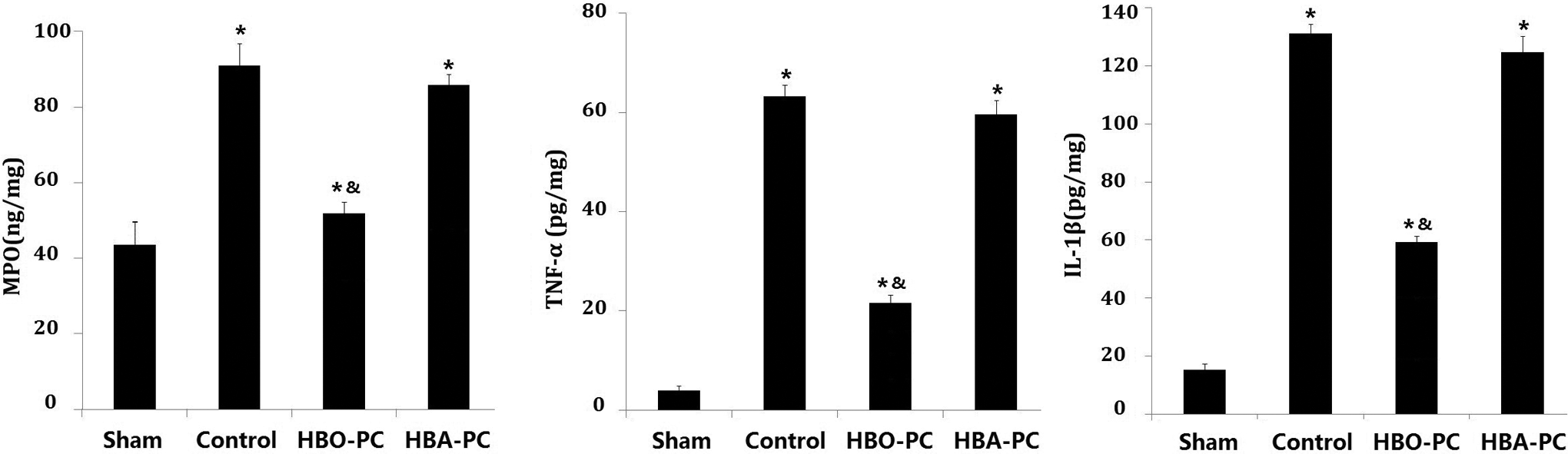
Myeloperoxidase (MPO), tumor necrosis factor α (TNF-α), and interleukin 1β (IL-1β) levels in myocardium tissues. Anti-inflammatory effects of hyperbaric oxygen preconditioning were evaluated by MPO, TNF-α, and IL-1β levels in myocardium tissues on the same day of cardiac functional measurement. Values are mean ± standard deviation (SD). *P < .05 compared to the Sham group and P < .05 compared to the Control group; n = 8 per group.
Protein Upregulation of HO-1 by HBO-PC and the Involvements of PI3K, Akt, and Nrf2 Signaling
Figure 5 shows the HBO-PC–induced HO-1 protein upregulation and the involvements of PI3K, Akt, and Nrf2 signaling by Western blot. As shown in Figure 5A and B, the expression of HO-1 was slightly increased by MIRI and further increased by HBO-PC. However, when the mice were treated by PI3K inhibitor LY294002, Nrf2-KO, or Akt inhibitor triciribine, the expression of HO-1 was greatly decreased. Figure 5A and C shows changes in the expression of Nrf2. The HBO-PC significantly enhanced expression of Nrf2. Similarly to HO-1, the expression of Nrf2 was also decreased by PI3K inhibitor LY294002 and Akt inhibitor triciribine. Figure 5A and D shows the phosphorylated Akt (p-Akt)/Akt changes. The HBO-PC greatly enhanced the phosphorylated Akt/Akt (p-Akt/Akt) value, which was decreased by PI3K inhibitor LY294002. The Nrf2-KO, however, did not affect the p-Akt/Akt value.

Representative Western blotting and densitometry of heme oxygenase 1 (HO-1), NF-E2-related factor 2 (Nrf2), and phosphorylated (p-Akt)/Akt. Representative Western blotting is shown in (A). Densitometry of HO-1, Nrf2, and phosphorylated Akt (p-Akt)/Akt is shown in (B)–(D), respectively. Mice were treated by phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002, Nrf2 knockout (Nrf2 KO), or Akt inhibitor triciribine before the ischemia-reperfusion procedure. Tissues were collected on the same day of cardiac functional measurement. Values are mean ± standard deviation (SD). *P < .05, n = 6 per group.
Changes in Cardiac Function by HO-1 PI3K, Akt Inhibitors, and Nrf2-KO
As shown in Figure 6, LVSP, +(dP/dt) max, and −(dP/dt) max were all decreased by HO-1 inhibitor ZnPP-IX, LY294002, Nrf2-KO, and triciribine (P < .05 compared to HBO-PC); LVDP was increased by ZnPP-IX, LY294002, Nrf2-KO, and triciribine (P < .05 compared to HBO-PC), but its value was still lower than that of Control (P < .05 compared to Control).

The changes in cardiac function by heme oxygenase 1 (HO-1), phosphatidylinositol 3-kinase (PI3K), and Akt inhibitor or Nrf2 knockout (Nrf2 KO). ± (dP/dt) max, left ventricular developed pressure (LVDP), and left ventricular systolic pressure were measured 24 days after the ischemia-reperfusion procedure. Mice were treated by HO-1 inhibitor zinc protoporphyrin IX (ZnPP-IX), PI3K inhibitor LY294002, Nrf2 KO, or Akt inhibitor triciribine. Results are expressed as mean ± standard deviation (SD; *P < .05 compared to HBO-PC and P < .05 compared to Control, n = 8).
Discussion
Our study confirmed that MIRI could significantly decrease cardiac functions and increase oxidative products including protein carbonyl, 8-OHdG, and MDA in the myocardium. The MIRI also leaded to inflammation, as shown by activation of MPO, TNF-α, and IL-1β. In the present study, all hemodynamic, biochemical, and histopathological results demonstrated the ability of HBO-PC to reduce the severity of MIRI in mice. Mice in the HBO-PC group exhibited dramatic improvement in cardiac functions, as manifested by increased LVSP and ±(dP/dt) max and decreased LVDP. The H&E staining showed that HBO-PC induced less necrosis, hemorrhage, neutrophilic infiltrate, and spindle-shaped interstitial cells. None of these amelioration, however, was shown in the HBA-PC group.
Many investigators have explored the effects of HBO-PC on myocardial infarction or MIRI, but the results are controversial. Radice et al exposed rats to 2.5 ATA HBO for 1, 3, or 6 hours, then removed the hearts and subjected them to low flow ischemia and reperfusion. 31 The results showed that the ischemic picture is worsened in hearts obtained from HBO-exposed animals. The left ventricular end-diastolic pressure (LVEDP) was increased significantly and proportionally according to the time of HBO exposure. Maffei et al subjected the isolated hearts from rats exposed to HBO to mild ischemia and reperfusion and found out that HBO greatly worsened the postischemic damage, as demonstrated by the rise in LVEDP and coronary perfusion pressure. 32 Han et al’s study, however, revealed that HBO preconditioning (2.5 HBO for 60 minutes, twice daily for 2 days) alleviated myocardial ischemia in rat model. The infarct size of the HBO-PC–treated group was significantly smaller; the heart function parameters including LVSP, +dP/dt max and −dP/dt max were significantly improved in the HBO-PC–treated group; the capillary density and VEGF protein levels were increased. 33 Cabigas et al found out that the HBO-PC decreased myocardial infarct size in isolated heart following I/R, in which NOS plays an essential role. 15 A clinical study demonstrated that HBO-PC prior to coronary artery bypass graft surgery caused an increase in left ventricular stroke work 24 hours following the surgery and the quantities of myocardial endothelial NOS and HSP72, indicating that HBO-PC could provide endogenous cardioprotection following ischemia-reperfusion injury. 34 The reason of the controversial outcomes of HBO-PC was not clear, which we speculate may involve the following: (1) the difference in HBO-PC protocol, including the pressure, duration, frequency of HBO, and the interval between HBO and I/R injury; (2) the difference in “ex vivo” experiment and “in vivo” experiment. Ex vivo experiment allows experimentation under more controlled conditions, while in vivo experiment is better suited for observing the overall effects on a living subject; and (3) the difference in experimental conditions and the animal species. Which reason was involved needs further clarification.
Biochemical assays showed the antioxidative ability of HBO-PC, as illustrated by decreased oxidative products; protein carbonyl; 8-OHdG; and MDA. The HBO-PC also showed its anti-inflammatory effect by suppression of MPO and proinflammatory cytokine TNF-α and IL-1β levels. The increase in oxidative products after MIRI indicated that ROS were produced by myocardium I/R procedure and contributed to the lethal MIRI. The HBO-PC, however, could decrease these oxidative products and protected myocardium from I/R injury. The inflammatory response and cytokines produced by myocardium I/R procedure play an important role in the pathophysiological process of MIRI. 35 Previous studies showed that acute inflammatory reaction regulated the I/R injury, 36 38 and the reduction in inflammatory cytokines or infiltration of leukocytes can attenuate MIRI. 39,40 Our results were consistent with these studies. The decrease in MPO, TNF-α, and IL-1β revealed that this could be one mechanism of the protective effects of HBO-PC.
Heme oxygenase-1 catalyzes the conversion of heme into biliverdin, carbon monoxide (CO), and free iron. 41 Many studies have shown that HO-1 plays a critical protective role in MIRI. 32 –45 Our results showed that the expression of HO-1 was slightly increased by MIRI and further increased by HBO-PC. The hemodynamic assays showed that HBO-PC-enhanced cardiac function was suppressed by HO-1 inhibitor ZnPP-IX, indicating the possible role of HO-1 in the effects of HBO-PC. The HO-1 may protect MIRI by the following mechanisms: (1) the degradation of heme. Free heme causes cell damage by direct attack or producing toxic ROS hydroxyl radicals through Fenton reaction and 46 (2) the catalytic byproduct CO. It is known that CO plays an important role in the cytoprotection against MIRI. 47 Exposure to CO is reported to protect MIRI by the activation of p38 mitogen-activated protein kinase, Akt, endothelial NOS, and cyclic guanosine onophosphate. 48
To explore the roles of Nrf2 and PI3K/Akt pathway in the protective effects of HBO-PC, we investigated the expression of HO-1, Nrf2, and p-Akt/Akt and the hemodynamic changes in the presence of their inhibitors. Our results showed that the expression of HO-1 was greatly decreased by PI3K inhibitor LY294002, Nrf2-KO, and Akt inhibitor triciribine. The expression of Nrf2 was enhanced by HBO-PC but significantly decreased by PI3K inhibitor LY294002 and Akt inhibitor triciribine. The HBO-PC greatly enhanced the p-Akt/Akt value, which was suppressed by LY294002 and triciribine, but not by Nrf2-KO. The underlying mechanism is not clear yet, but we speculate that in the signal pathway in which Akt, PI3K, and Nrf2 were involved, the Akt was at the upstream position, so the Akt can regulate expression of Nrf2, but Nrf2 cannot regulate the expression of Akt. The hemodynamic assays showed that HBO-PC-enhanced cardiac function was suppressed by HO-1 inhibitor ZnPP-IX, LY294002, Nrf2-KO, and triciribine, indicating the roles of HO-1, Nrf2, PI3K, and Akt in the effects of HBO-PC. Taken together with the results from the inhibitor studies, these data strongly present a novel signaling mechanism by which HBO-PC protects MIRI via PI3K/Akt/Nrf2-dependent antioxidant defensive system. However, we should notice that a 2-fold induction of HO-1 occurs following I/R injury and a 4-fold induction occurs with both I/R injury and HBO-PC. Either, more HO-1 is required for cytoprotection or HO-1 is required prior to injury to elicit protection, but the inhibition of the Akt-Nrf2 mechanism reduced HO-1 induction by only 2-fold. Therefore, it is unclear whether the mechanism limits both the induction from preconditioning and injury by 2-fold, or whether the mechanism favors either the preconditioning or injury. Further examination is therefore warranted to determine these distinctions.
NF-E2-related factor-2, a member of the Cap “n” Collar basic-leucine-zipper family of transcription factors, is a key transcriptional factor for ARE-regulated genes which protect against the toxic effects of ROS. 49 It could regulate the expression of phase-II detoxification and antioxidant enzymes under oxidative stress, including HO-1. Our results showed that under the mild oxidative stress induced by HBO-PC, the expression of both Nrf2 and HO-1 was enhanced. In Nrf2-knockout mice, however, the HBO-PC failed to induce the expression of Nrf2 and HO-1, inducing the deterioration of the cardiac function. The role of Nrf2 in the HO-1 induction and the effect of HBO-PC were displayed by these results.
The involvement of PI3K/AKT pathway in the induction of Nrf2/ARE was also investigated in the present study. We found that PI3K/Akt pathways were activated after HBO-PC and the treatment with LY294002 significantly resulted in a decrease in expression of p-Akt, Nrf2, and HO-1 after HBO-PC. Thus, we propose that PI3K pathway is required for expression of Nrf2/HO-1. Next, we observed the Akt inhibitor triciribine significantly decreased the expression after HBO-PC, demonstrating that PI3K/AKT signaling pathway is involved in expression of HO-1. These observations are consistent with previous reports that PI3K/Akt signaling pathway is implicated in Nrf2/HO-1 induction. Wang’s study showed that activities of both the basal and induced Nrf2 were inhibited by PI3K inhibitors wortmannin and LY294002, which also inhibited sulforaphane-induced Nrf2 nuclear translocation, while overexpression of Akt stimulated Nrf2 activation. 50 Raju et al found out that raw garlic homogenate could reduce myocardial oxidative stress through PI3K/Akt/Nrf2-Keap1 dependent pathway, 51 whereby increased ratio of p-Akt/Akt might activate Nrf2’s antioxidant activity and protect heart from oxidative stress. Evaluation of signaling pathways by Deng et al 52 showed that LY294002 significantly suppressed PM2.5-induced Nrf2 nuclear translocation and messenger RNA expression of HO-1, indicating the involvement of PI3K/Akt in Nrf2-mediated HO-1 transcription. In another study, however, the authors found out that the pretreatment of endothelial cells with LY294002 increased Nrf2 and HO-1 protein and decreased ARE-luciferase activity. 26 The reason for the different role of PI3K/Akt pathway in the Nrf2/ARE induction is not yet clear, but it indicated that under some circumstance, the PI3K/Akt pathway may suppress the expression of Nrf2/ARE. The mechanism whereby PI3K/Akt may stimulate expression of Nrf2/HO-1 is less clarified but may involve peroxynitrite which plays a role in activation of Nrf2 and Nrf2 binding to the ARE via the pathway of PI3K. 53 Protein kinase C (PKC) is believed to play a role in formation of peroxynitrite that activates Nrf2. 54 It is also reported that the rearrangement of actin microfilaments and the rise in cellular Ca2+, which is necessary for nuclear translocation of Nrf2, were involved. 55 Further studies are required to clarify the specific mechanism in the MIRI setting.
Conclusion
Hyperbaric oxygen preconditioning protected the cardiac functional and histological changes induced by MIRI, decreased oxidative injury and inflammation. A novel signaling mechanism was revealed by which HBO-PC protects MIRI via PI3K/Akt/Nrf2-dependent antioxidant defensive system.
Footnotes
Author Contributions
Yin, X contributed to conception and design, contributed to analysis, drafted the manuscript, critically revised the manuscript, gave final approval, and agree to be accountable for all aspects of work ensuring integrity and accuracy; Zhao, K contributed to conception and design, critically revised the manuscript, gave final approval, and agreed to be accountable for all aspects of work ensuring integrity and accuracy; Wang, X contributed to acquisition and analysis, drafted the manuscript; Fan, Z contributed to acquisition and interpretation; Peng, C contributed to interpretation; Ren, Z contributed to design; Huang, L contributed to analysis.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.