
Abstract
The sarcolemmal adenosine triphosphate (ATP)-sensitive K+ (sarcKATP) channel in the heart is a hetero-octamer comprising the pore-forming subunit Kir6.2 and the regulatory subunit sulfonylurea receptor SUR2A. By functional analysis of genetically engineered mice lacking sarcKATP channels, the pathophysiological roles of the K+ channel in the heart have been extensively evaluated. Although mitochondrial KATP (mitoKATP) channel is proposed to be an important effector for the protection of ischemic myocardium and the inhibition of ischemia/reperfusion-induced ventricular arrhythmias, the molecular identity of mitoKATP channel has not been established. Although selective sarcKATP-channel blockers can prevent ischemia/reperfusion-induced ventricular arrhythmias by inhibiting the action potential shortening in the acute phase, the drugs may aggravate the ischemic damages due to intracellular Ca2+ overload. The sarcKATP channel is also mandatory for optimal adaptation to hemodynamic stress such as sympathetic activation. Dysfunction of mutated sarcKATP channels in atrial cells may lead to electrical instability and atrial fibrillation. Recently, it has been proposed that the gain-of-function mutation of cardiac Kir6.1 channel can be a pathogenic substrate for J wave syndromes, a cause of idiopathic ventricular fibrillation as early repolarization syndrome or Brugada syndrome, whereas loss of function of the channel mutations can underlie sudden infant death syndrome. However, precise role of Kir6.1 channels in cardiac cells remains to be defined and further study may be needed to clarify the role of Kir6.1 channel in the heart.
Keywords
Introduction
It is already 30 years since Noma for the first time discovered adenosine triphosphate (ATP)-sensitive K+ (KATP) channel in the cardiac cells 1 . It has been clarified that KATP channel is a hetero-octamer comprising 2 subunits: the pore-forming subunit having 2 membrane-spanning regions (Kir6.1 or Kir6.2) and the regulatory subunit sulfonylurea receptor (SUR1, SUR2A, or SUR2B; Figure 1). 2,3 Functional analysis of genetically engineered mice lacking KATP channels has enabled us to understand the pathophysiological roles of the K+ channel in the cardiovascular system. 4,5 Our functional study using Kir6.2 knockout mice indicated that sarcolemmal KATP (sarcKATP) channel in cardiac cells plays an important role in cardioprotection against ischemia/reperfusion injury by ischemic preconditioning. 6 However, it has been postulated that mitochondrial KATP (mitoKATP) channel rather than sarcKATP channel is important for the protection of ischemic myocardium. 7 Although the molecular identity of mitoKATP channels is currently unknown, a recent report 8 has suggested that renal outer medullary potassium channel (ROMK; Kir1.1) may be a pore-forming subunit of mitoKATP channel. Dysfunction of KATP channel may become an arrhythmogenic factor. Mutations in sarcKATP channels may lead to increased susceptibility to atrial fibrillation (AF). 9 In addition, a gain-of-function mutation in sarcKATP channel with Kir6.1 pore subunit has been suggested to be a cause of J wave syndrome. 10 In this review, the role of KATP channels in cardiac arrhythmias is discussed.

Schematic presentation of sarcKATP channel and mitoKATP channel and their roles in cardiac cells. SarcKATP channel is hetero-octamer comprising the pore-forming subunit Kir6.2 and the regulatory subunit sulfonylurea receptor SUR2A. The sulfonylurea receptor has 2 NBFs (NBF-1 and NBF-2) on the cytoplasmic side. Kir6.2 is a member of the inward rectifier K+ channel family having 2 transmembrane domains. K+ channel openers such as pinacidil and nicorandil activate sarcKATP channel. HMR 1883 selectively and glibenclamide nonselectively block sarcKATP channel. Although not established, mitoKATP channel on inner mitochondrial membrane may be composed of ROMK and short-splicing variant of SUR2. 8,56 K+ channel openers such as diazoxide, nicorandil, and pinacidil activate mitoKATP channel. 5-Hydroxydecanoate and glibenclamide are selective and nonselective blockers of mitoKATP channel respectively. Endogenous substances such as norepinephrine, adenosine, and bradykinin activate their G protein-coupled receptors, phospholipase C–protein kinase C pathway, and mitoKATP channel, thereby producing “ischemic preconditioning.” However, sarcKATP channel is also important for the establishment of ischemic preconditioning. 6 sarcKATP indicates sarcolemmal adenosine triphosphate-sensitive K+; mitoKATP, mitochondrial KATP; NBFs, nucleotide-binding folds; ROMK, renal outer medullary potassium channel.
ATP-Sensitive K+ Channels and Ischemia/Reperfusion-Induced Arrhythmias
Noma suggested that activation of KATP channels is responsible for action potential shortening observed under hypoxic or ischemic conditions when he found the KATP channel in cardiac cells for the first time. 1 Since KATP channel openings are supposed to produce cardioprotective effect against ischemic injuries, 11 many pharmaceutical companies tried to develop KATP channel activators (K+ channel openers). Nicorandil, a K+ channel opener clinically available, was shown to activate not only sarcKATP channel but also mitoKATP channel (Figure 1). 12,13 In the late 1990s, much attention has been focused on mitoKATP channel rather than sarcKATP channel as an effector for cardioprotection. 14,15 However, functional molecules of the mitoKATP channel have not been identified with certainty. In our previous studies, 6,16 the function of mitoKATP channel, indirectly evaluated by flavoprotein oxidation, was preserved in ventricular cells isolated from Kir6.2- and Kir6.1-knockout mice. Therefore, it is unlikely that Kir6.1 or Kir6.2 is a pore molecule of mitoKATP channel. Recently, Foster et al have proposed that ROMK (Kir1.1) may be a molecular component of mitoKATP channel (Figure 1). 8 Further studies may be needed to test this hypothesis.
Since ischemia/reperfusion-induced ventricular arrhythmias are sometimes lethal, medical prevention of the malignant ventricular arrhythmias is extremely important. In anesthetized rabbits, nicorandil and bimakalim were reported to suppress ventricular arrhythmias during ischemia/reperfusion, concomitantly with reduction in infarct size. 17 Das and Sarkar 18 suggested that the antiarrhythmic and cardioprotective effects of K+ channel openers could be ascribed to activation of mitoKATP channels rather than sarcKATP channels.
Several studies indicated that selective inhibition of sarcKATP channels can inhibit ischemia/reperfusion-induced arrhythmias and improve survival rates of experimental animals. 19 –21 Pharmaceutical companies tried to develop selective sarcKATP-channel blockers but this type of drug is not clinically available at the present time. Why can the selective sarcKATP-channel blocker not be used in clinical settings? From the results of our functional study using Kir6.2-null mice, 22 we suggested that activation of sarcKATP channels contributes to the action potential shortening but it is not the primary cause of extracellular K+ accumulation in the ischemic myocardium. In the ischemic myocardium of Kir6.2-knockout mice, action potential duration (APD) was rather prolonged concomitantly with prolongation of effective refractory period (ERP). In addition, the ischemia-induced conduction delay was more marked in the myocardium of Kir6.2-knockout mice than that in the myocardium of wild-type (WT) mice. Since there were no differences in the magnitude of extracellular K+ accumulation and in the decreases in the resting membrane potential between the ventricular tissues of WT and Kir6.2 mice, the greater conduction delay in the ischemic myocardium of Kir6.2-kockout mice might be ascribed to decreased conductance of gap junctions, probably resulting from excessive intracellular Ca2+ overload. Lack of action potential shortening in the ischemic myocardium of Kir6.2-knockout mice might increase Ca2+ influx during plateau phase and produce intracellular Ca2+ overload, resulting in cell-to-cell uncoupling and mechanical dysfunction. 6,22 Thus, selective sarcKATP-channel blocker may prevent ischemia/reperfusion-induced ventricular arrhythmias in the acute phase. However, treatment with a selective sarcKATP-channel blocker may aggravate the ischemic damages and may be associated with a poor long-term prognosis.
ATP-Sensitive K+ Channels and Arrhythmias in Stressed or Failing Hearts
Terzic and his colleagues evaluated the role of sarcKATP channels in the adaptation to a variety of hemodynamic stress. 23 –26 The Kir6.2-knockout mice showed decreased tolerance to a treadmill exercise stress test. 23 The Kir6.2-knockout mice easily developed ventricular arrhythmias concomitantly with prolongation of QT interval in electrocardiogram (ECG) and cardiac death in response to catecholamine injection. 23,24 In addition, the Kir6.2-null mice developed heart failure and maladaptive remodeling in response to experimental hypertension and aortic constriction. 25,26 These findings suggest that sarcKATP channel is mandatory for optimal adaptation to physiological or pathophysiological stress. When the heart was exposed to hemodynamic stress, sarcKATP channels might be activated and regulate the APD, thereby preventing intracellular Ca2+ overload due to exaggerated Ca2+ influx during plateau phase.
Sarcolemmal ATP K+ channels in the heart may be activated even under physiological conditions with increased metabolic demand. Sympathetic activation increases heart rate and myocardial contractility, resulting in enhanced oxygen demand. Relatively anaerobic energy production in the heart with increased oxygen demand and sympathetic stimulation during vigorous exercise accelerate glycolysis and intracellular lactate accumulation. It was reported that intracellular lactate activated sarcKATP channels in the presence of moderate levels of ATP in rabbit ventricular cells. 27 Although APD shortening associated with adrenergic stimulation has been mainly ascribed to an increase in slow component of the delayed rectifier K+ current (I Ks), 28 activation of sarcKATP channels may also contribute to the APD shortening. Thus, sarcKATP channels may be activated, and cardiac action potentials may be shortened during strenuous exercise and sympathetic activation in humans (Figure 2). Action potential shortening due to sarcKATP channel activation is expected to reduce the time for Ca2+ influx via the L-type Ca2+ channels during the plateau phase and to increase the time for Ca2+ extrusion through the Na+–Ca2+ exchange system (Figure 2). Sarcolemmal ATP-sensitive K+ channels may be extremely important for the adaptation to physiological stress such as sympathetic stimulation and vigorous exercise. In the heart of marathon runners, sarcKATP channels may be activated and protect the cardiomyocytes from intracellular Ca2+ overload.

Adaptation of the heart to vigorous exercise and sympathetic stimulation with activation of sarcKATP channels. In the heart of healthy individuals at rest, action potential repolarization in the ventricle is mainly regulated by I Kr and I Ks. During strenuous exercise and sympathetic stimulation, heart rate increases and action potentials are shortened by enhancement of I Ks. Activation of sarcKATP channels may also contribute to the action potential shortening during vigorous exercise and sympathetic stimulation. Action potential shortening due to increases in I K.ATP and I Ks can reduce the time for Ca2+ influx via the LTCCs during plateau phase and increase the time for Ca2+ extrusion through the NCX during diastolic intervals. Such adaptive responses are important for prevention of intracellular Ca2+ overload in cardiac cells. sarcKATP indicates sarcolemmal adenosine triphosphate-sensitive K+; I Kr, rapid component of the delayed rectifier K+ current; I Ks, slow component of the delayed rectifier K+ current; I K.ATP, adenosine triphosphate-sensitive potassium current; LTCC, L-type Ca2+ channels; NCX, Na+–Ca2+ exchange system.
It was reported that the sensitivity to the K+ channel opener diazoxide was increased in atrial and ventricular cells of human hearts with congestive heart failure. 29 Greater sensitivity to the K+ channel opener may partly explain increased susceptibility to AF in the failing heart. Changes in sarcKATP channel function in diseased state may be important for the induction of cardiac arrhythmias.
Repetitive episodes of ventricular fibrillation (VF) were reported to produce APD shortening that was ascribed to activation of sarcKATP channels. 30 –32 Shortened action potentials can easily induce recurrent VF in failing hearts. Blockade of sarcKATP channels by glibenclamide or HMR 1883 was shown to promote spontaneous defibrillation, increase the rate of successful defibrillation, and prevent the recurrence of VF. 30,32 However, treatment with sarcKATP-channel blockers for a prolonged time may aggravate myocardial damages by intracellular Ca2+ overload, as discussed previously. Therefore, it would be appropriate to use sarcKATP-channel blockers only for a limited time.
ATP-Sensitive K+ Channels and AF
Some of familial atrial fibrillation (AF) is known to be associated with mutations in K+ channel genes. A gain-of-function mutation in KCNQ1, which encodes the pore-forming α subunit of I Ks channel, was associated with familial AF. 33 In addition, gain-of-function mutations in K+ channel genes, which encode pore-forming proteins of ultrarapid delayed rectifier K+ current (I Kur) 34 and transient outward current (I to), 35 were also reported to enhance AF susceptibility.
In terms of sarcKATP channel, a mutation in ABCC9 gene encoding regulatory SUR2 subunit was reported to confer predisposition to paroxysmal AF. 36 Blunted response of the mutated SUR2A to adenosine diphosphate was suggested to lead to electrical instability with adrenergic AF. The study also indicated that Kir6.2-knockout mice easily developed AF following isoproterenol injection, which was in contrast to WT mice maintaining sinus rhythm even after sympathomimetic challenge. 36 These findings suggest that dysfunction of sarcKATP channels can produce intracellular Ca2+ overload in atrial cells and be a predisposition to AF under adrenergic stress. It is noteworthy that high-frequency focal activity in the pulmonary veins, which is important for the initiation and perpetuation of AF, can also be enhanced by intracellular Ca2+ overload. 37 In this context, loss-of-function mutation in KCNA5 gene encoding Kv1.5 channel, through which I Kur flows, was associated with idiopathic AF. 38 Thus, both gain-of-function and loss-of-function mutations in cardiac K+ channels can lead to increased susceptibility to AF.
Long-term AF can produce electrical remodeling in atrial tissues. It was reported that shortening of ERP at more than 3 hours after rapid atrial pacing was observed in dogs and HMR1098, a selective sarcKATP-channel blocker, partially antagonized the ERP shortening 39 suggesting that sarcKATP channels might be activated several hours after induction of AF. However, it was reported that in atrial cells from patients with persistent AF the density of ATP-sensitive potassium current and messenger RNA (mRNA) level of Kir6.2 were reduced. 40,41 The reduced density of sarcKATP channels in atrial cells of persistent AF might cause intracellular Ca2+ overload, leading to further progression of electrical remodeling and enhanced focal activity.
ATP-Sensitive K+ Channels and J Wave Syndrome
Recently, much attention has been focused on J wave syndrome as a cause of idiopathic VF (IVF). An early repolarization (ER) pattern in the ECG, characterized by J-point elevation, slurring of the terminal part of the QRS and ST-segment elevation, has long been considered to be a benign ECG manifestation that is more commonly seen in young healthy individuals and athletes. 42 However, mounting evidence that the ER pattern can be associated with a risk of VF has been provided. 43 Therefore, ER syndrome (ERS) cannot be always considered to be benign. In the ECG of patients with Brugada syndrome (BrS), an accentuated J wave imitating incomplete right bundle branch block (RBBB) and ST-segment elevation in the right precordial leads were found. 44,45 Accordingly, Antzelevitch has proposed that ERS and BrS represent a continuous spectrum as J wave syndrome because they share similar ECG characteristics, clinical outcomes, risk factors, and underlying electrophysiological abnormalities (Figure 3). 46
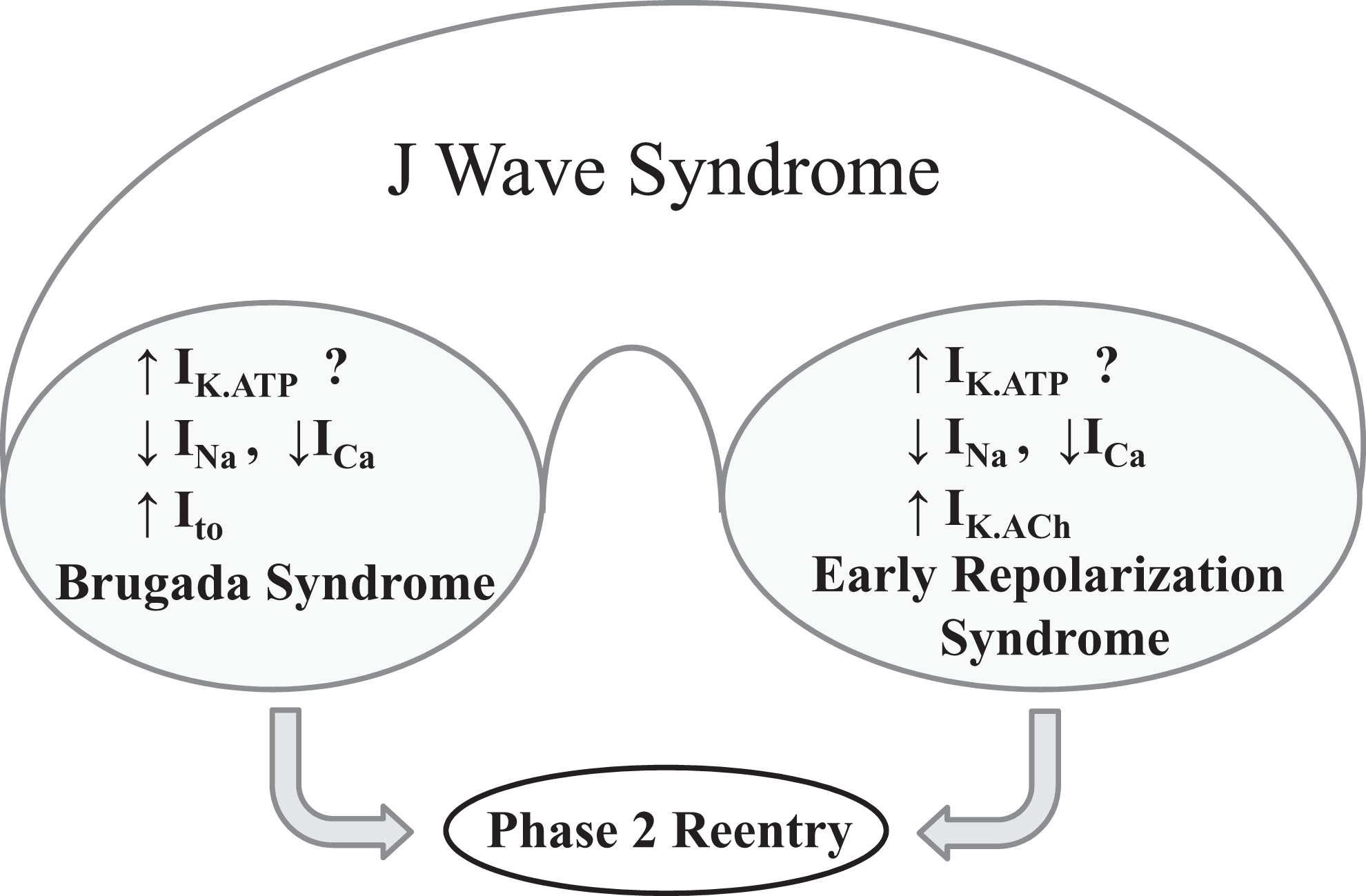
Schematic depicting J wave syndrome. Antzelevitch hypothesized that a decrease in Na+ or Ca2+ current or an increase in K+ current such as I to and I K.ATP can give rise to accentuated J waves-associated ERS and BrS. Both syndromes can be triggered by closely coupled phase 2 reentrant extrasystoles. Modified with permission from Antzelevitch. 10 Ito indicates transient outward current; I K.ATP, adenosine triphosphate-sensitive potassium current; ERS, early repolarization syndrome; BrS, Brugada syndrome.
Based on the available clinical data, ERS has been classified into 3 types for risk stratification for patients with ER. 46 –48 An ER pattern manifest exclusively in the lateral precordial leads has been designated as type 1; ER pattern in the inferior or inferior-lateral leads has been designated as type 2; and an ER pattern appearing globally in the inferior, lateral, and right precordial leads has been labeled type 3. In addition, ERS as well as BrS has been subclassified into BrS1-BrS12 and ERS1-ERS6 based on genetic ion-channel mutations. 46 Genetic analysis revealed that variable mutations in Na+, Ca2+, and K+ channel genes were found in patients with BrS and ERS. Among them a mutation in KATP channel gene was reported in patients with ERS and BrS. In 2009, Haïssaguerre et al reported a case with ERS, showing missense mutation (S422L) in KCNJ8 gene encoding Kir6.1 α subunit of KATP channel. 49 It was also reported that the same missense mutation was identified in 3 BrS and 1 ERS proband. 50 In addition, the same mutation in KCNJ8 gene was detected in 2 of 325 lone AF probands and 1 of 2 cases showed ER pattern on the ECG. 51 Functional analysis of KCNJ8-S422L revealed that the KATP current of Kir6.1-S422L mutation was significantly increased compared to Kir6.1-WT channels. 52 Therefore, it is assumed that the gain-of-function mutation of cardiac KATP channel Kir6.1 may be a pathogenic substrate for J wave syndromes. On the other hand, it has been suggested that loss-of-function mutations in KCNJ8 can underlie sudden infant death syndrome (SIDS). Tester et al performed mutational analysis of KCNJ8 on genomic DNA isolated from necropsy tissue of 292 unrelated patients with SIDS. 53 They identified 2 novel KCNJ8 mutations (E332del and V346I), both of which localized to Kir6.1’s C-terminus and showed that they were loss-of-function mutations. Thus, it has been suggested that gain-of-function mutations of Kir6.1 may lead to J wave syndrome whereas loss-of-function mutations may be involved in SIDS.
In terms of SIDS resulting from loss-of-function mutations in KCNJ8 gene, dysfunction of coronary arteries secondary to Kir6.1 mutations may explain the sudden death in infants, because Kir6.1 protein is essential for KATP channel function in vascular smooth muscle cells. 54 Indeed, we previously found sudden death with Prinzmetal angina and atrioventricular block in Kir6.1-deficient mice. 16 Can the gain-of-function mutations in KCNJ8 gene encoding Kir6.1 α subunit of KATP channel lead to ERS or BrS? Although both Kir6.1 and Kir6.2 mRNA were abundantly expressed in hearts of WT mice, 54 the density of sarcKATP current and response to the K+ channel opener pinacidil in ventricular cells of Kir6.1 knockout mice were not different from those in ventricular cells of WT mice. 16 If the findings observed in mice can be extrapolated to humans, it is unlikely that the gain-of-function mutations in KCNJ8 gene can underlie J wave syndrome. However, there may be differences in the functional roles of Kir6.1 proteins in the heart between rodents and humans. Further study may be needed to clarify the role of Kir6.1 channel in the heart.
Sarcolemmal ATP-sensitive K+ channels function as a metabolic sensor and couple cellular metabolism to electrical activity in many organs, maintaining their Ca2+ and energy homeostasis. 4 Recently, there have been many reports suggesting causal relationship between mutations in the subunit genes of KATP channels and cardiac arrhythmias. 55 Especially, much attention has been focused on role of mutated Kir6.1 channel in J wave syndrome. However, precise role of Kir6.1 channels in cardiac cells remains to be defined. Functional studies to elucidate the role of Kir6.1 channel in cardiac cells are warranted.
Footnotes
Acknowledgments
The authors especially thank Prof S. Seino, Prof T. Miki, and collaborators involved in the studies from our laboratory, which are cited in this review. We are also grateful to Ms Y. Reien, Mr H. Maruyama, and Ms T. Tachibana for their technical assistance and Ms I. Sakashita for her secretarial assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports, and Culture of Japan [Grant 23390053].