
Abstract
Atherogenesis has been traditionally viewed as a metabolic disease representing arterial obstruction by fatty deposits in its wall. Today, it is believed that atherogenesis involves highly specific biochemical and molecular responses with constant interactions between various cellular players. Despite the presence of inflammatory reaction in each and every step of atherosclerosis from its inception to terminal manifestation, the cause–effect relationship of these 2 processes remains unclear. In this article, we have attempted to review the role of inflammation in the development of atherosclerosis and in its major complication—coronary heart disease.
Introduction
Coronary artery disease (CAD) is a major cause of death in the United States, accounting for nearly 20% of all US deaths. 1 In terms of absolute numbers, 1.5 million individuals develop acute myocardial infarction (AMI) and more than 500 000 die every year, which means about 2 heart attacks every minute. Yet there has been almost 50% reduction in mortality from AMI over the last 30 years. Reduction in mortality reflects better understanding of the process of AMI and improved treatment (both medical and surgical) of this troublesome complication of the atherosclerotic process. There have also been major strides in our understanding of the pathogenesis of atherosclerosis and underlying risk factors, such as smoking, diabetes mellitus, hypertension, and dyslipidemia.
Atherosclerosis was previously thought to be a mere pipeline obstruction by fatty deposits in the arterial wall. Over the few decades, many facets of the process leading to atherosclerosis have been unraveled. Today we believe that atherogenesis involves highly specific cellular and molecular responses with constant interactions between various cellular players. There is recognition of inflammatory reaction in the atherosclerotic region from beginning of the fatty streak to culmination into an acute event resulting from plaque erosion or rupture. Although the association of inflammation with AMI has been known for over 50 years, growing evidence suggests that inflammation influences plaque progression and its vulnerability to rupture. 2 However, it is not clear whether inflammation and atherogenesis represent a cause and effect relationship, or inflammation is a response to tissue injury.
Here, we review the role of inflammation in the process of development of atherosclerosis and its relevance with particular reference to CAD.
Molecular and Cellular Dynamics in Atherosclerosis
Experimental and clinical studies have shown that under the influence of well-known risk factors, such as smoking, hypertension, diabetes, and dyslipidemia, endothelial cells become dysfunctional, develop pores in interendothelial junctions, and exhibit signs of injury (Figure 1). The injured/dysfunctional endothelium develops a surface that is conducive to inflammatory cells adhesion, rolling, and migration to subendothelial region. This presumably marks the initiation of atherosclerosis.
3
The 3 major consequences of such injury are as follows: A cartoon illustration of steps involved in the process of atherogenesis. In the presence of traditional risk factors, such as diabetes, smoking, hypertension, and dyslipidemia, the endothelium becomes activated leading to the expression of adhesion molecules. Monocytes attach to the activated endothelium and migrate to subendothelial space where they engulf oxidized LDL (ox-LDL) and transform into macrophages and foam cells. Release of growth factors, such as angiotensin-II (Ang II) leads to fibroblast and smooth muscle cell (SMC) growth and migration. Oxidized LDL is taken up by endothelial cells via specific scavenger receptors (SRs), such as lectin-like oxidized LDL receptor 1 (LOX-1). Oxidized LDL in small concentrations induces angiogenesis and releases metalloproteinases (MMPs), causing platelet aggregation. Oxidized LDL also stimulates the expression of Ang II type 1 receptors (AT1Rs). Both ox-LDL and Ang II reduce endothelial nitric oxide (NO) synthase activity, resulting in diminished NO release, and also stimulate the formation of superoxide anions (O2
−) and other free radicals. The earliest lesion in atherosclerosis, the “fatty streak,” appears in susceptible individuals at a very young age. This lesion consists of cells (monocyte-derived macrophages, macrophage-derived foam cells, and T lymphocytes) filled with lipids. The subsequent growth of the plaque is characterized by a chronic inflammatory fibroproliferative response as seen in other chronic disease states such as cirrhosis, rheumatoid arthritis, and pulmonary fibrosis. Inflammatory cells (monocytes and T cells) adhere and then roll along the activated endothelium as a result of upregulation of adhesion molecules on endothelium. These cells become activated and rapidly migrate through gaps in the endothelial lining to the subendothelial region. Three families of adhesion molecules are considered key players in leukocyte–endothelial adhesion, namely, selectins, integrins, and members of the immunoglobulin superfamily of proteins.
4,5
The role of different adhesion molecules is summarized in Table 1. The movement of leukocytes from the bloodstream to the vessel wall is a highly organized process and occurs in several distinct steps.
6
The importance of these inflammatory cells in atherogenesis is illustrated by the observation that there is virtual absence of atherosclerosis when macrophage-colony-stimulating factor (M-CSF) or P-selectin null genotype is introduced in murine models of severe dyslipidemia.
7,8
Platelets play an important role in the proinflammatory milieu. Although formerly thought to be passive players in atherogenesis, it is now evident that these cells are important not only in thrombosis resulting in acute ischemic event but also in perpetuating the inflammatory environment. Platelets secrete several vasoactive chemokines and cytokines (e.g. CD40L, thrombospondin, platelet-activating factor), regulated on activation, normal T cell expressed and secreted (RANTES), extractable nuclear antigen ENA-78, macrophage inflammatory protein (MIP), and chemokine (C-X-C motif) ligand 4 (CXCL4) with autocrine and paracrine effects.
9
Activated platelets attach to leukocytes, resulting in the formation of platelet–leukocyte aggregates.
10
Thromboxane A2, a potent proaggregatory eicosanoid released from activated platelets, is also a proinflammatory stimulus. Further, free cholesterol and oxidized low-density lipoprotein cholesterol (ox-LDL), major components of the plaque, are potent proinflammatory stimuli triggering the recruitment of more macrophages.
11
Oxidized LDL itself stimulates platelet aggregation in response to ADP and thrombin most likely by activation of lectin-like oxidized LDL receptor 1 (LOX-1) on platelets.
12
Therefore, platelets play multiple roles in atherothrombosis (Figure 2). The third major consequence of the endothelial activation/injury/dysfunction is abnormal vasomotor function. Normal healthy endothelium produces nitric oxide (NO) from 
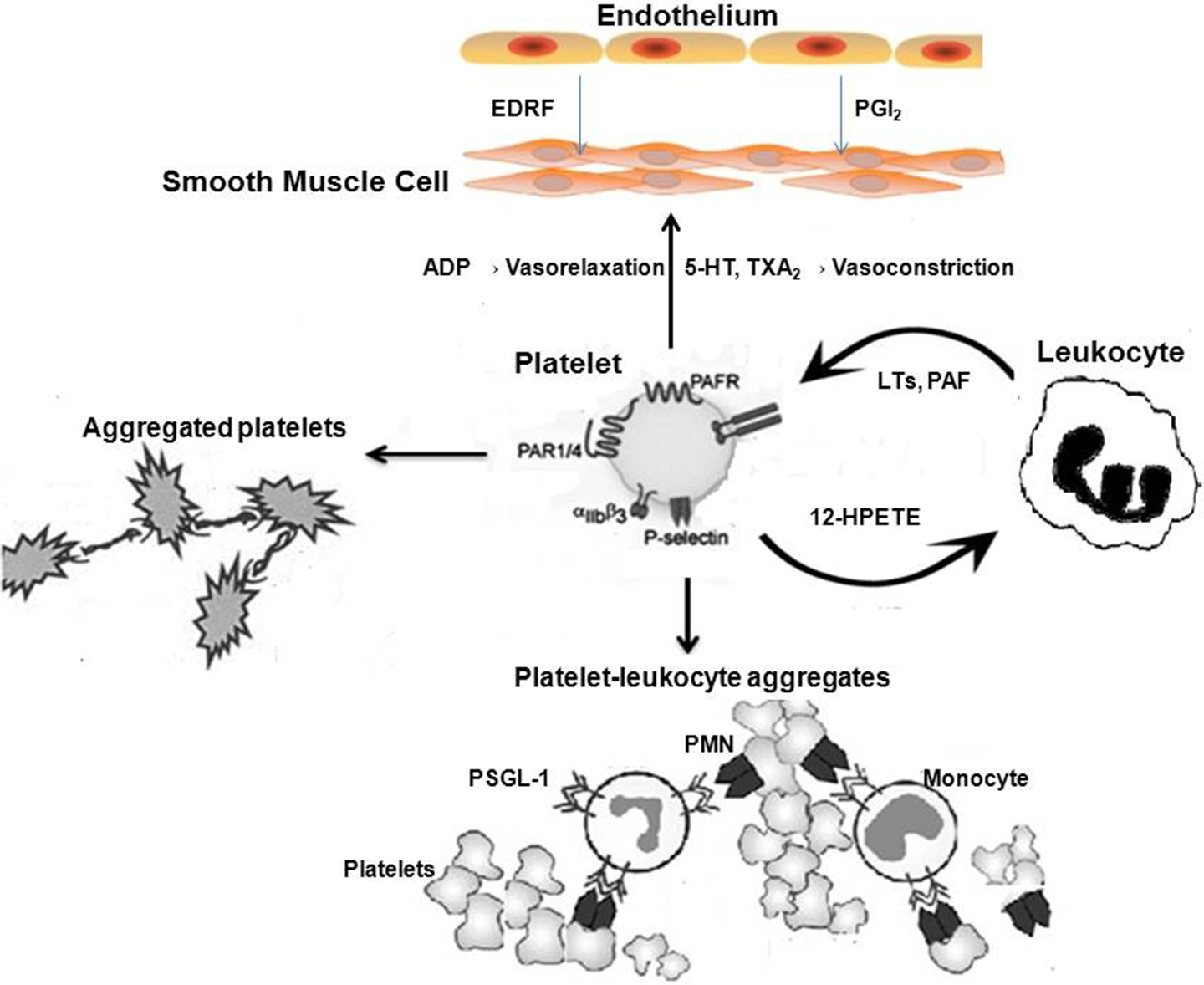
Cartoon demonstrating the inflammatory steps involving leukocytes and platelets. Soon after vessel wall injury/inflammation, platelets adhere to endothelium, followed by leukocytes attaching both to platelets and directly to endothelial cells. Initial interaction between leukocyte–platelet is mediated by P-selectin (on platelets) and PSGL on leukocytes. This results in the release of chemokines from platelets like IL-8, RANTES, PDGF, and ENA-78, all causing leukocyte intergrin activation, adhesion between leucocyte integrin and platelet surface molecules, and a procoagulant state through activation of extrinsic and intrinsic clotting pathway. ADP indicates adenosine diphosphate; EDRF, endothelium-derived relaxing factor; 5-HT, 5-hydroxytryptamine; HPETE, hydroxyperoxytetraenoic acid; PMN, polymorphonuclear leukocyte; PSGL, P-selectin glycoprotein ligand; PAF, platelet-activating factor; PAF, platelet activating factor receptor; TxA2, thromboxane A2; LTs, leukotrienes.
Important Molecules Involved in Leukocyte Endothelial Interactions.

Inflammation and Oxidative Stress
The inflammatory response itself can influence lipoprotein transfer within the vessel wall. 15 Proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin 1 (IL-1), M-CSF, increase the binding of LDL to endothelium and SMCs and increase the transcription of the LDL-receptor gene. 16 There is also evidence regarding the release of large amounts of ROS in all stages of atherosclerosis. Reactive oxygen species induce endothelial dysfunction by denaturing NO, act as chemoattractants for inflammatory cells, activate platelets, induce SMC proliferation, and oxidize proatherogenic lipids. 17,18 Activated leukocytes also release ROS, oxidizing LDL cholesterol, which is taken up by scavenger receptors on endothelium, SMCs, and monocytes/macrophages. After binding to scavenger receptors, ox-LDL initiates a series of intracellular events that include the induction of proteases, proinflammatory cytokines, and cellular apoptosis. Thus, a vicious cycle of inflammation, modification of lipoproteins, and further inflammation can be maintained in the artery by the presence of modified lipoproteins. In short, inflammation begets inflammation and the cycle continues. Activated monocytes/macrophages engulf ox-LDL and transform into foam cells. It is the deposition of lipid-laden foam cells in the subendothelial space that gives the appearance of fatty streak. The complex interaction between inflammation and ROS is summarized in Figure 3.

Cartoon illustrating the reactive oxygen species (ROS)-inflammation interaction in atherosclerosis. The ROS cause accumulation of monocytes in vessel wall, endothelial dysfunction, platelet aggregation, and vascular smooth muscle cell (VSMC) proliferation and migration. Monocytes in the vessel wall form macrophages and elicit proinflammatory cytokines and chemokines, which cause influx of lipids into various cells including macrophages that form foam cells. These lipid molecules in turn trigger additional ROS formation, perpetuating this vicious cycle, leading to the formation of atherosclerotic plaque. Ox-LDL, oxidized LDL; mm-LDL, minimally modified LDL; ROS, reactive oxygen species; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; MCP-1, monocyte chemoattractant protein 1; VEGF, vascular endothelial growth factor; MMP, matrix metalloproteinase; IL, interleukin; TNF, tumor necrosis factor; eNOS, endothelial nitric oxide.
Time Course of Monocyte/Macrophage Accumulation in the Arterial Wall
Marwali et al 19 studied the accumulation of CD68+ cells (monocytes/macrophages) over the course of atherosclerosis in LDLr-deficient mice given high cholesterol diet and observed that accumulation of CD68+ cells in the aorta increased progressively over the first 10 weeks of lipid deposition in the arterial wall. Thereafter, a significant decline in the number of these cells in the vessel wall was observed; however, the atherosclerotic lesion continued to grow. Mitra et al 20 have shown that after initial exposure to ox-LDL, the expression and activity of adhesion molecule gene decreased during subsequent passage of cells, which in turn may account for the decrease in the accumulation of inflammatory cells in mature lesions. Nonetheless, it is evident that the rupture-prone regions show erosion or rupture of cap lining the plaque and sparse SMCs, but intense accumulation of foam cells filled with ox-LDL as well as immune cells in the underlying region. 21
Role of Immunity in Atherosclerosis
The presence of immune cells and immune cell products (such as the chemokines that were discussed earlier) in atherosclerotic lesions indicates the participation of systemic and local vascular immune system in atherogenesis. There are 2 arms of immunity, the innate immunity, which is rapid and nonspecific, and the adaptive arm, which is slow but more precise. Pathogens and autoantigens activate these 2 arms to cause vascular inflammation. Activation and differentiation of circulating immune cells have been linked with clinical manifestations of atherosclerosis. 22 There is compelling evidence from mouse models of atherosclerosis that knocking out monocyte chemotaxis mediators like monocyte chemoattractant protein 1 (MCP-1) and C Chemokine receptor 2 (CCR2) interferon-γ or its receptor, costimulatory molecules, CD40L, toll-like receptors (TLRs), and the recombination activating gene (RAG) genes, all lead to a reduction in atherosclerosis. 23 On the other hand, knocking out IL-10, a Th1 inhibitory cytokine, resulted in increase in such lesions. 24 The clinical evidence of higher likelihood of CAD among patients with autoimmune diseases like lupus and rheumatoid arthritis further supports the role of immunity in atherosclerosis. 25
Inflammatory Markers in CAD
Work from several laboratories suggests that the degree of inflammation correlates with the severity and outcome of patients with myocardial ischemia. Others have suggested that inflammation markers may predict development of disease in asymptomatic individuals. Various markers of inflammation have been studied; some of which are in use in clinical setting.
C-reactive protein (CRP) has been the most widely studied in relation to presence of inflammation, coronary artery pathology, and coronary heart disease outcome. It is an acute-phase reactant synthesized in liver in response to IL-6 and is elevated in all inflammation-associated disease states. Once ligand-bound, CRP can activate the classical compliment pathway, stimulate phagocytosis, and bind to immunoglobulin receptors. A number of investigators have confirmed the increased risk in acute coronary syndrome (ACS) associated with high concentrations of CRP, 26 –29 above and beyond troponin levels. More importantly, CRP has been reported to have prognostic value even among patients with negative cardiac troponin and no evidence of myocyte necrosis. 26 –28 On the other hand, a number of investigators have refuted these observations. 30 –32 Methodological issues have also been highlighted, and the independence between CRP and troponin release questioned. 33 Despite much promise in the early years, hs-CRP as a risk assessment tool and as a target for therapy in individual patients has not found much use in general practice.
Myeloperoxidase (MPO) is a peroxidase enzyme that is abundantly expressed in polymorphonuclear cells (neutrophils) and plays an important role in neutrophil microbicidal action. The MPO causes oxidative modification of LDL cholesterol that is a key event in the development and progression of atherogenesis. Two recent studies have shown that a single measurement of plasma MPO at hospital admission predicted the risk of major adverse cardiac events in the subsequent 30-day and 6-month periods. 34,35 Further studies on MPO in patients with and without CAD are needed to determine its sensitivity and specificity.
Simple rise in white blood cell (WBC) count has been associated with adverse clinical outcomes and a high mortality rate in the setting of ACS. 36,37 Madjid et al 38 looked at the value of WBC count versus other sophisticated proinflammatory biomarkers and showed that WBC count is as or more valuable in predicting CAD events. With its simplicity and widespread availability, WBC seems an attractive marker. However, the nonspecific nature of this marker significantly limits its use.
Serum amyloid A (SAA) is yet another important acute-phase protein. Like its acute-phase counterpart hsCRP, SAA may provide important prognostic information with respect to short-term mortality among patients presenting with an ACS. 39 While a prognostic role for hsCRP in unstable ischemic heart disease has now been demonstrated in prospective studies, further studies are needed to validate SAA in ACS and further define its sensitivity and specificity.
Monocyte chemoattractant protein 1 is a potent chemotactic factor for monocytes. It is responsible for recruitment of monocytes to the sites of inflammation, which appears to be critical in promoting plaque instability. 40 In case–control studies, plasma MCP-1 concentrations were shown to be associated with restenosis after percutaneous transluminal coronary angioplasty, suggesting the importance of inflammation in the pathogenesis of restenosis. 41 Nonetheless, the distribution of MCP-1 values in the healthy participants and the study population overlapped considerably in one prospective study; thus limiting the overall value of this protein as a biomarker. 42
Lipoprotein-associated phospholipase A2 (Lp-PLA2) is a calcium independent phospholipase A2 enzyme secreted by leukocytes and associated with circulating LDL and macrophages in atherosclerotic plaques. The Lp-PLA2 generates 2 proinflammatory mediators, lysophosphatidylcholine and oxidized nonesterified fatty acids, which play a major role in the formation of atherosclerotic lesions. There is growing evidence from primary prevention studies as well as secondary prevention studies that this inflammatory marker may be an independent predictor of cardiovascular events. 43 Evidence also points toward its role as a marker of endothelial dysfunction. 44
Circulating Vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), IL-6, and TNF-α levels have all been evaluated but the data are conflicting and nonspecific. 45
Anti-Inflammatory Agents in the Management of CAD
Although many forms of anti-inflammatory therapies targeted at blocking inflammatory response in atherosclerosis have been demonstrated to be useful in mouse models 46,47 their role in humans has not been established. Similar to various cell adhesion molecules like ICAM and VCAM, MCP-1 are expressed on the surface of endothelial cells after activation during atherogenesis and their blockade with monoclonal antibody has been shown to retard atherogenesis. 46 Similarly, adhesion molecules like integrins play a role in cell adhesion, migration, cell–matrix interaction in atherosclerosis; and blockade of integrins has been shown to induce a stable plaque phenotype in mouse models. 47
In humans, trials have been conducted with potent anti-inflammatory drugs like steroids and other less potent drugs like nonsteroidal anti-inflammatory drugs. These agents failed to reduce the burden of atherosclerosis in human studies. 48 Aspirin, on the other hand, has been found to be consistently useful in secondary prevention of CAD-related events and possibly in primary prevention in high-risk individuals. 49 Some herbs (turmeric, ginger, and garlic) and natural plants such as green tea have also been proposed to have anti-inflammatory effect, but as yet there are no definitive clinical trials with these agents. 50,51
Succinobucol (AGI-1067) is a novel anti-inflammatory agent that can reduce inflammation in the walls of blood vessels and provide antioxidant protection (vascular protectants). In a study aggressive reduction of inflammation stops events (ARISE), designed to assess the benefits of succinobucol over current standard of care in patients with CAD, no difference was found in the primary end point, which was a composite of cardiovascular death, cardiac arrest, MI, stroke, unstable angina, and coronary revascularization. There was, however, a significant 19% reduction in cardiovascular death, cardiac arrest, MI, and stroke (secondary end point), and 64% significant reduction in new onset diabetes. 52 Very interestingly, this agent increased LDL cholesterol and decreased high-density lipoprotein (HDL) cholesterol.
Statin group of drugs may exert an anti-inflammatory effect (the so-called pleiotropic effect). Animal studies showed that statins may reduce the activity of nuclear factor-κB (NF-κB), a transcription factor controlling proinflammatory genes, and expression of cyclooxygenase 2 (COX-2) in models of atherosclerosis. 53 The first indication that such an effect may be clinically relevant came in late 1990s from Cholesterol And Recurrent Event trial (CARE) in which the reduction in recurrent MI associated with pravastatin was found to be significantly greater among those with higher hsCRP levels, an effect that was present irrespective of the baseline levels of total cholesterol, HDL-C, and LDL-C. 54 Recent data from 2 clinical trials suggested that the level of CRP achieved as a result of using statin therapy might be as important as the level of LDL-C achieved with use of these agents. 55,56 In a study by Nissen et al, 56 the CRP level was independently and significantly correlated with the rate of progression of atherosclerosis, which is determined by intravascular ultrasonography. However, the major effect of this class of drugs is LDL-cholesterol lowering. As discussed earlier, free cholesterol and ox-LDL are proinflammatory mediators, and lowering their levels with statins is the primary basis of reduction in CRP and other inflammatory markers.
There is some evidence that angiotensin-converting enzyme (ACE) inhibitors may exert anti-inflammatory effects. Studies indicate angiotensin-II enhances the synthesis and release of IL-6, an effect that can be blocked by inhibiting ACE. 57 As IL-6 mediates B-Lymphocyte maturation, complement activation and cytokine release, ACE inhibitors may exert anti-inflammatory effect by suppressing these proinflammatory cytokines. However, the major effect of ACE inhibitors is modulation of renin–angiotensin system activity.
Aspirin is known for many years to be a potent platelet aggregation inhibitor through the blockage of COX-1 pathway. In recent years, it has been suggested that anti-inflammatory actions of aspirin are quite important, and the antiplatelet and anti-inflammatory actions are interlinked and act in conjunction to exert beneficial effect on CAD events. 58 In studies designed to study the effect of aspirin in primary prevention of CAD, it was shown that risk reduction attributable to aspirin is greater among individuals with elevated CRP than in those with lower CRP. 59 The cardioprotective effect of aspirin in the prevention of CAD events is unique to aspirin and cannot be generalized to all COX inhibitors such as ibuprofen or indomethacin.
Thiazolidinediones (TZDs) are oral insulin sensitizers that enhance glucose uptake by skeletal muscle and are developed to improve glycemic control in diabetic patients. They are ligands for the nuclear receptor peroxisome proliferator activator receptor-gamma, a member of the nuclear receptor superfamily that functions as a key transcriptional regulator of cell differentiation. The TZDs block vascular smooth muscle cell growth by inducing cell cycle arrest in G1 phase. These agents inhibit the expression of VCAM-1 and ICAM-1 in activated endothelial cells and significantly decrease the homing of macrophages and monocytes to atherosclerotic plaques. 60 Once again, like COX-2 inhibitors, a statistically significant increase in MI in patients taking rosiglitazone (a prototype of TZD) was seen in a meta-analysis of 16 observational studies. 61
To date, there is no direct evidence to suggest that this beneficial effect of statins, renin angiotensin system (RAS) inhibitors, and TZDs is due to the countering of the inflammatory process. The quest to assess the effect of inflammation suppression in atherosclerosis progression has interested drug companies and investigators to develop and test the efficacy of a direct approach like use of low-dose methotrexate, canakinumab (the high-affinity human monoclonal, antihuman IL-1β), and darapladib (Lp-PLA2 inhibitor). The results of these clinical trials may provide more insight into the relationship of inflammation and atherosclerosis. 62 –64
Targets Beyond Direct Modulation of Inflammation
In the pathogenesis of atherosclerosis, scavenger receptors are involved in the lipid transport mechanisms. Although most of them had been described on the macrophages, few candidate receptors have been demonstrated in endothelial cells like class A scavenger receptor with C-type lectin (SRCL or CLP-1), the class B scavenger receptor (CD36), and class E scavenger receptor LOX-1. LOX-1 a has gained interest over the past decade for its association with atherogenesis. LOX-1 is constitutively present in endothelial cells and its expression can be induced by shear stress, ROS, endothelin 1, angiotensin-II, advanced glycation end products and, interestingly, by ox-LDL itself. 65,66 It is responsible for the internalization, endocytosis, and degradation of ox-LDL in macrophages that leads to foam cell formation. 67 Oxidized LDL is the major ligand for LOX-1. It also serves as a binding receptor to many other ligands including modified HDL, CRP, heat shock proteins, activated platelets, apoptotic cells, bacteria, phosphatidylserine and other phospholipids. Oxidized LDL through LOX-1 activates arginase-II that causes downregulation of endothelial NO synthase leading to endothelial dysfunction. 68 There is strong evidence to suggest LOX-1 as a powerful inflammatory mediator. 69 LOX-1, through the NF-κB pathway, increases vascular smooth muscle cell proliferation and migration, a key feature of intimal hyperplasia. Recently, LOX-1 deletion was shown to reduce inflammation, ROS generation, and atherosclerosis in LDLr-null mice given high cholesterol diet. 70 In keeping with in vitro and in vivo work, serum levels of soluble LOX-1, a posttranslational product of LOX-1 have been shown to be upregulated in patients with ACS. 71 Another molecule, LOX-1 ligand containing apoB (LAB), was shown to predict the risk of stroke and adverse cardiovascular events in a prospective cohort of patients followed for 11 years. Notably, drugs like statins and ACE inhibitors have been shown to affect LOX-1-mediated pathways in vitro. 72
Inflammation: a Response to Injury or a Primary Mediator of Atherosclerosis?
Atherosclerosis is a complicated process with multifactorial etiology and involves inflammatory, fibroproliferative, and angiogenic responses. Despite the presence of inflammatory cells in the atherosclerotic tissues and in the plasma of patients with CAD from beginning of the disease process to precipitation of an acute vascular event, the cause–effect relationship remains unclear. The aforementioned association of various inflammatory markers in atherosclerosis does not naturally imply that inflammation is the primary mediator of atherosclerosis.
In 19th century, Robert Koch laid out a postulate to establish causal relationship of disease with the offending agent. According to the 3 components of this postulate, the causal agent must be isolated from the diseased participant or region; the disease must be cured when the agent is removed by some means, and the disease should reappear on reinoculation of the agent. In case of inflammation and atherosclerosis, although there is strong evidence that atherosclerosis and related disorders are associated with elevated levels of inflammatory biomarkers, there is no evidence to support that reduction of inflammation- or induction of inflammation-inducing agent/agents will reduce the cardiovascular events. Hence, the loop of causality remains unclosed at this time. Novel approaches are underway to investigate this relationship further.
Over the years, it has become clear that atherosclerosis is a multifactorial disease in which there are many associations like the inflammatory process. A major challenge in atherogenesis is how to identify specific stage of atherosclerosis and knowing what targets to use in that specific stage rather than a shotgun approach. This may allow a more pathogenesis-driven therapy and provide more plausible insights into the pathophysiology of the disease.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.